


Редкий дефицит фактора свертывания крови
Бюллетень WHEC Практика и клинической управления для медицинских работников. Образование гранта, предоставленного здоровья женщин и образовательный центр (WHEC).
Clotting factors are proteins in the blood that control bleeding. When a blood vessel is injured, the walls of the small blood cells contract to limit the flow of blood to the damaged area. Then, small blood cells called platelets stick to the site of injury and spread along the surface of the blood vessel to stop the bleeding. At the same time, chemical signals are released from small sacs inside the platelets that attract other cells to the area and make them clump together to form what is called a platelet plug. On the surface these activated platelets, many different clotting factors work together in a series of complex chemical reactions (known as the coagulation cascade) to form a fibrin clot. The clot acts like a mesh to stop bleeding. Coagulation factors circulate in the blood in an inactive form. When a blood vessel is injured, the coagulation cascade is initiated, and each coagulation factor is activated in a specific order to lead to the formation of the blood clot. Coagulation factors are identified with Roman numerals (e.g. factor I or FI). Rare clotting factor deficiencies are a group of inherited bleeding disorders caused by a problem with one or several clotting factors. Problems with factor VIII and factor IX are known as hemophilia A and B, respectively.
The purpose of this document is to identify rare clotting factor deficiencies and management. Rare clotting factor deficiencies are bleeding disorders in which one of the other factors (i.e. factor I, II, V, V + VIII, VII, X, XI, or XIII) is missing or not working properly. Less is known about these disorders because they are diagnosed so rarely. In fact, many have only been discovered in the last 40 years.
Prevalence
By definition, rare factor deficiencies have a prevalence of less than 200,000 in the US population, or an incidence of less than 1 in 2,000 in Europe (1). The very small numbers of patients with rare clotting factor disorders present challenges in diagnosis, evaluation of bleeding risk and treatment. Use of new assays, full genome sequencing, and global clotting assays will significantly improve diagnosis of patients with rare bleeding disorders.
Characteristics of Rare Clotting Factor Deficiencies
Clinical symptoms among patients of rare clotting factor deficiencies vary significantly between disorders, and patients, even when affected with the same disorder. Heterozygous individuals commonly do not manifest a bleeding tendency. Mucocutaneous and surgical associated bleeding were reported in 20% patients, whereas post-traumatic hemarthrosis and hematomas are rarely reported in FVII and FX deficiencies. Menorrhagia, spontaneous abortion, and bleeding during vaginal delivery were reported in ~20% of women with all deficiencies (2).
Women with rare clotting factor deficiencies require specific attention and care; in addition to the common bleeding symptoms, they may also experience gynecologic bleeding (3). Beyond menorrhagia, affected females are at increased risk of hemorrhagic cysts, endometriosis, endometrial hyperplasia, polyps, and fibroids. Pregnancy and childbirth pose particular clinical challenges, and miscarriages, bleeding during pregnancy and postpartum hemorrhage have been frequently reported in some deficiencies. These events impact quality of life and employment.
Rare Bleeding Disorder Distribution Worldwide
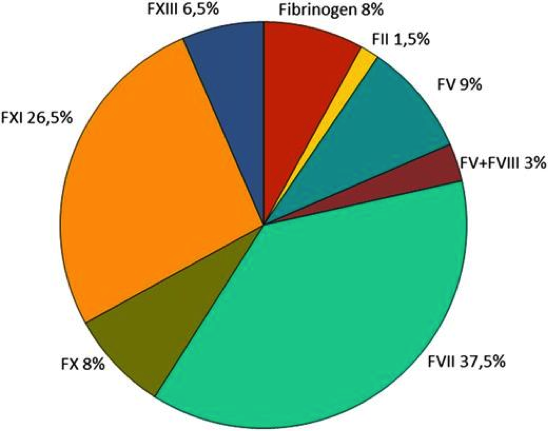
Figure 1. Worldwide distribution of rare bleeding disorders (RBD) derived from the World Federation of Hemophilia (WFH) and European Network of the Rare Bleeding Disorders (EN-RBD)
Clotting factors are proteins in the blood that control bleeding. Many different clotting factors work together in a series of chemical reactions to stop bleeding. This is called the clotting process. Hemophilia A and B, are problems with factor VIII and factor IX.
Classification
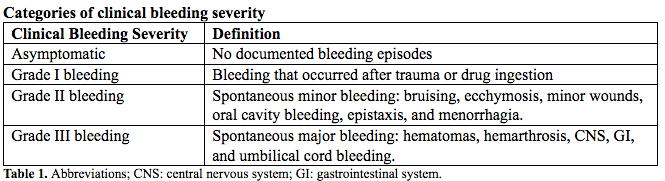
Clinical bleeding episodes are classified into four severity categories based on the location, potential clinical impact, and bleeding trigger either being spontaneous, after trauma, or drug induced (See Table 1 below). This study documented a strong association between residual coagulant activity and clinical bleeding severity for deficiencies of fibrinogen, combined FV + VIII, FX, and FXIII, with a weak association for FV and FVII deficiencies; residual FXI activity did not predict clinical bleeding severity (4). From the same study, it was documented that the minimum level to ensure complete absence of clinical symptoms is different for each disorder. Thus, rare bleeding disorders cannot be considered as a single class of disorders; instead, studies should focus on evaluation of specific aspects of each individual rare bleeding disorders.
Each rare bleeding disorders can have several bleeding symptoms ranging from minor post-traumatic to severe episodes of appearing at birth or later in life. In some deficiencies, residual coagulant activity is directly related to the hemorrhagic risk, yet this is not true for all.
Overview of Specific Rare Clotting Factors Disorders
Fibrinogen Deficiency (Factor I or FI)
Fibrinogen deficiency is heterogenous, with two main phenotypes distinguished: plasma and platelet protein levels are not measurable or very low, leading to afibrinogenemia and hypofibrinogenemia, whereas low clottable fibrinogen with normal or moderately reduced fibrinogen antigen results in dys- and hypodysfibrinogenemia (5). Afibrinogenemia is an autosomal recessive disorder, which means that both parents must carry the defective gene in order to pass it on to their child. Like all autosomal recessive disorders, afibrinogenemia is found more frequently in areas of the world where marriage between close relatives is common. Hypofibrinogenemia, dysfibrinogenemia, and hypodysfibrinogenemia can be either recessive (both parents carry the gene) or dominant (only one parent carries and transmits the gene). All types of factor I deficiency affect both males and females.
Diagnosis: Factor (FI) deficiency is diagnosed by a variety of blood tests, including a specific test that measures the amount of fibrinogen in the blood. However, low fibrinogen levels or abnormal function may be a sign of another disease, such as liver or kidney disorders, which should be ruled out before a bleeding disorder is diagnosed.
Treatment: there are three treatments available for factor I deficiency (6). All are made from human plasma. 1) Fibrinogen concentrate, 2) Cryoprecipitate, and 3) Fresh Frozen Plasma (FFP).
Prothrombin Deficiency (factor II or FII)
Prothrombin deficiency is the rarest inherited coagulation disorder with a prevalence of ~1 in 2 million (7). Prothrombin, a vitamin K-dependent glycoprotein that is hepatically synthesized, is the zymogen of the serine protease α-thrombin and is encoded by a gene of -21 kb located on chromosome 11 (8).
Factor II deficiency is an autosomal recessive disorder, which means that both parents must carry the defective gene in order to pass it on to their child. It also means that the disorder affects both males and females. It can also be acquired later in life as a result of liver disease, vitamin K deficiency, or certain medication such as the blood-thinning drug Coumadin®. Acquired factor II deficiency is more common than the inherited form.
Diagnosis: Factor II deficiency is diagnosed by a variety of blood tests. The doctor will need to measure the amount of factors II, V, VII, and X in the blood. Diagnostic tests should be performed by a specialist.
Treatment: there are two treatments for factor II deficiency. Both are made from human plasma. 1) Prothrombin complex concentrates (PCCs), 2) Fresh Frozen Plasma (FFP).
Factor V Deficiency (FV deficiency)
FV has a dual role in coagulation: it is a protein cofactor required by the prothrombinase complex for thrombin generation and contributes to the proteins C/S anticoagulant pathway by downregulating FVIII activity (9). Severe deficiency typically presents early in life; nonetheless FV deficiency is clinically heterogeneous, as despite lower FV levels, severe patients may not bleed as expected. Megakaryocytes can synthesize FV; however, the majority of platelet FV is endocytosed from plasma. Platelet degranulation and release of platelet FV at the site of vascular injury is a critical contributor to local FV concentration. Furthermore, there is evidence that platelet FV that is locally released in high concentrations is less susceptible to inhibition, supporting normal hemostasis.
Factor V deficiency and inherited bleeding disorder that is caused by a problem with factor V. it is and autosomal recessive disorder, which means that both parents must carry the defective gene in order to pass it on to their child. It also means the disorder affects both males and females. Factor V deficiency is very rare, but like all autosomal recessive disorders, it is found more frequently in areas of the world where marriage between close relatives is common.
Diagnosis: People with abnormal levels of factor V should also have their factor VIII levels checked to rule out combined factor V and factor VIII deficiency, which is completely separate disorder.
Treatment: Factor V deficiency is usually only needed for severe bleeds or before surgery. FFP is the usual treatment because there is no concentrate containing only factor V. Platelet transfusions, which contain factor V, are also sometimes an option.
Combined Factor V and Factor VIII deficiency
Combined FV and FVIII deficiency (F5F8D) is characterized by concomitantly low levels (usually 5-20%) of both coagulant activity and antigen (10). Interestingly, the concomitant deficiency of these two coagulation factors does not enhance the hemorrhagic tendency observed in each separate defect. The combined deficiency is completely separate from factor V deficiency and factor VIII deficiency (hemophilia A).
Combined factor V and VIII deficiency is an autosomal recessive disorder, which means that both parents must carry the defective gene in order to pass it on to their child. It also means that the disorder affects both males and females. The deficiency is very rare, but like all autosomal recessive disorders, it is found more frequently in areas of the world where marriage between close relatives is common. Most cases are found around the Mediterranean Sea, especially in Israel, Iran and Italy. Normally the disorder is caused by a single gene defect the body's ability to transport factor V and factor VIII outside the cell into blood stream, and not by a problem with the gene for either factor.
Diagnosis: Combined factor V and factor VIII deficiency is diagnosed by a variety of blood tests to determine if the levels of both factors are lower than normal. The specialists should perform test.
Treatment: there are three treatments available for combined factor V and factor VIII deficiency.
1) Factor VIII concentrate; 2) Fresh Frozen Plasma (FFP); and 3) Desmopressin.
Factor VII Deficiency (FVII deficiency)
Factor VII deficiency is the most common autosomal recessive coagulation disorder, 1 in 500,000 and is typically clinically heterogeneous, ranging in severity from lethal to mild, or even asymptomatic (11). It is an autosomal recessive disorder, which means both parents must carry the defective gene in order to pass it on to their child. It also means that the disorder affects both males and females. Factor VII deficiency may be inherited with other factor deficiencies. It can also be acquired later in life as a result of liver disease, vitamin K deficiency, or certain medications such as the blood-thinning drug Coumadin®
Diagnosis: Factor VII deficiency is diagnosed by a variety of blood test that should be performed by a specialist.
Treatment: there are several treatments available for factor VII deficiency, these are: 1) Recombinant VIIa concentrate (rFVIIa), 2) Factor VII concentrate, 3) Prothrombin complex concentrate (PCC) containing factor VII, and 4) Fresh Frozen Plasma (FFP).
Factor X Deficiency (FX deficiency)
FX is a glycoprotein pivotal to the coagulation cascade, as the first enzyme in the common pathway of thrombin formation. FX is hepatically synthesized and encoded by the FX gene (F10), comprising 22 kb and located on chromosome 13, a few kilobases downstream of the F7 gene (12). FX deficiency is an inherited bleeding disorder that is caused by a problem with factor X. because the body produces less FX than it should, or because the FX is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
FX deficiency is an autosomal recessive disorder, which means that both parents must carry the defective gene in order to pass it on to their child. It also means that the disorder affects both males and females. FX deficiency is one of the rarest inherited clotting disorders, but like all autosomal recessive disorders, it is found more frequently in areas of the world where marriage between close relatives is common.
Diagnosis: FX deficiency is diagnosed by a variety of blood tests that should be performed by a specialist at a hemophilia/bleeding disorders treatment center.
Treatment: There are two treatments available for FX deficiency. Both are made from human plasma. 1) Prothrombin complex concentrate (PCC) containing factor X, 2) Fresh Frozen Plasma (FFP).
Factor XI Deficiency (FXI deficiency)
The estimated prevalence of severe FXI deficiency in most populations is ~1 in 1 million, but it is higher in Ashkenazi Jews where heterozygosity approaches 8% (13). The relationship between plasma FXI levels and the bleeding tendency is not as clear-cut as in other rare bleeding disorders. Bleeding phenotype is not correlated with genotype but rather than site of injury. Injury in an area of high fibrinolytic activity (e.g., urogenital tract, oral cavity after dental extraction, or tonsillectomy) increases bleeding risk about 49% to 67% compared with sites with less fibrinolytic activity about 1.5% to 40% (14).
Factor XI deficiency is an inherited bleeding disorder, also called hemophilia C. It differs from hemophilia A or B in that there is no bleeding into joints and muscles. Factor XI deficiency is the most common of the rare bleeding disorders and the second most common bleeding disorder affecting women, after von Willebrand disease. Some people have inherited factor XI deficiency when only one parent carries the gene. The disorder is most common among Ashkenazi Jews, that is, Jews of Eastern European ancestry.
Diagnosis: Factor XI deficiency is diagnosed by a variety of blood tests that should be performed by a specialist at a hemophilia/bleeding disorder treatment center.
Treatment: There are several treatments available to help control bleeding in people with factor XI deficiency. These are: 1) Factor XI concentrate, 2) Antifibrinolytic drugs, 3) Fibrin glue, and 4) Fresh Frozen Plasma (FFP).
Factor XIII Deficiency (FXIII deficiency)
FXIII deficiency is a transglutaminase functioning to cross-link the α and γ fibrin chains, resulting in increased clot strength and fibrinolytic resistance (15). Patients with FXIII-A deficiency have a bleeding tendency that is usually severe, with early onset of life-threatening symptoms (e.g., umbilical cord and CNS bleeding) in up to 80% and 30% respectively (16).
Factor XIII deficiency is an inherited bleeding disorder. It is an autosomal recessive disorder, which means that both parents must carry the defective gene in order to pass it on to their child. It also means that the disorder affects both males and females. Factor XIII deficiency is very rare, but like all autosomal recessive disorders, it is found more frequently in areas of the world where marriage between close relatives is common.
Diagnosis: Factor XIII deficiency is difficult to diagnose. Standard blood clotting tests do not detect the deficiency, and many laboratories are not equipped with more specialized tests that measure the amount of factor XIII in a blood sample or how well factor XIII is working. The high rate of bleeding at birth usually leads to early diagnosis.
Treatment: There are several treatments available to help control bleeding in people with factor XIII deficiency. These are: 1) Factor XIII concentrate, 2) Cryoprecipitate, and 3) Fresh Frozen Plasma (FFP). A new recombinant FXIII-A2 concentrate (rFXIII-A2) is available, and a phase 3 clinical trial that was recently completed demonstrated that rFXIII is safe and effective in bleed prevention in congenital FXIII-A subunit deficient patients (17). rFXIII has recently been approved for the treatment of FXIII-A deficiency in Australia, Canada, the European Union, Switzerland, and the United States.
Combined deficiency of vitamin K-dependent clotting factors (VKCFD)
Inherited combined deficiency of the vitamin K-dependent clotting factors (VKCFD) is a very rare inherited bleeding disorder that is caused by a problem with clotting factors II, VII, IX and X (18). In order to continue the chain reaction of the coagulation cascade, these four need to be activated in a chemical reaction that involves vitamin K. When this reaction does not happen the way it should, the clotting reaction is blocked, and the blood clot does not form.
VKCFD is an autosomal recessive disorder, which means that both parents must carry the defective gene in order to pass it on the child. It also means that the disorder affects both males and females. VKCFD can also be acquired later in life as a result of disorders of the bowel, liver disease, dietary vitamin K deficiency, or certain medications such as the blood-thinning Coumadin®. Acquired VKFCD is more common than the inherited form. Some newborn babies have a temporary vitamin K deficiency, which can be treated with supplements at birth.
VKCFD commonly presents early in life with intracranial hemorrhage or umbilical stump bleeding; routine vitamin K administration may delay neonatal diagnosis. Severely affected children may present with skeletal abnormalities including nasal and distal digital hypoplasia, epiphyseal stippling, and mild conductive hearing loss. Older patients can present with easy bruising and mucocutaneous or postsurgical bleeding.
Diagnosis: VKCFD is diagnosed by a variety of blood tests that should be performed by a specialist at a hemophilia/bleeding disorders treatment center. Care should be taken, particularly in newborns, to exclude causes of acquired vitamin K deficiency or exposure certain medications.
Treatment: Oral or parenteral vitamin K1 should promptly started. Some patients demonstrate inadequate response. Limited data exist on the effectiveness of 10 mg vitamin K1 weekly prophylaxis. Massive parenteral doses do not always correct activity levels with persistence of undercarboxylated molecules; here factor replacement via PCC or alternatively a virally inactivated FFP could be used in acute bleeding episodes or prior to surgery.
There are three treatments available for VKCFD: 1) Vitamin K, 2) Prothrombin complex concentrate (PCC), and 3) Fresh Frozen Plasma (FFP).
Global Hemostasis Tests
Standard coagulation screen tests reveal the basic integrity of the coagulation process; evaluation of coagulation speed and extent are limited by test sensitivity at very low residual factor levels. Tests evaluating global hemostatic capacity (thrombin generation test and thromboelastography) may provide more accurate evaluation of in vivo hemostasis and treatment response and be better suited to predict clinical phenotype as they more effectively assess rate/total thrombin generated, whole blood clot formation, and/or fibrin polymerization. Recently, these tests have been used to evaluate hemostasis in patients with rare bleeding disorders, specifically FV and FXI deficiency (19).
Global Hemostasis Tests
Standard coagulation screen tests reveal the basic integrity of the coagulation process; evaluation of coagulation speed and extent are limited by test sensitivity at very low residual factor levels. Tests evaluating global hemostatic capacity (thrombin generation test and thromboelastography) may provide more accurate evaluation of in vivo hemostasis and treatment response and be better suited to predict clinical phenotype as they more effectively assess rate/total thrombin generated, whole blood clot formation, and/or fibrin polymerization. Recently, these tests have been used to evaluate hemostasis in patients with rare bleeding disorders, specifically FV and FXI deficiency (19).
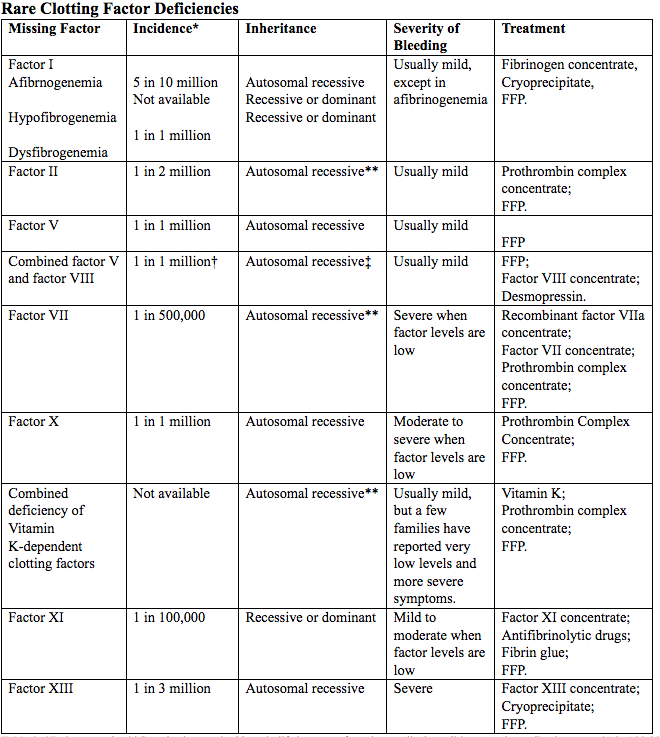
Table 2. *Estimates only; **Can also be acquired later in life because of another medical condition, certain medications etc.; †1 in 100,000 in some populations, including Israel, Iran, and Italy; ‡Very rarely, factor VIII deficiency can be inherited separately from only one parent.
Abbreviated: FFP (Fresh Frozen Plasma)
Source: World Federation of Hemophilia
These assays represent an emerging strategy to determine therapeutic effectiveness and to monitor rare bleeding disorders treatment, particularly FXI deficiency where standard assays fail to correlate with bleeding risk (20). Test standardization to reproduce reliable thrombin generation measurements facilitated by standardized preanalytical and analytical procedures is required before widespread clinical use.
Molecular Diagnosis
Molecular diagnosis is based on causative mutation identification in genes encoding corresponding coagulation factor (21). Exceptions are combined FV and FVIII deficiency, caused by mutations in genes encoding proteins involved in FV and FVIII intracellular transport (MCFD2 and LMAN1) and VKCFD, caused by mutations in genes encoding enzymes involved in in post-translational modification and vitamin K metabolism (GGCX and VKOR). Inheritance pattern is autosomal recessive for all rare bleeding disorders, except for some cases of FXI and hypo- and dysfibrinogenemia. Missense mutations are most frequent, representing 50% to 80% of identified mutations, except for LMAN1 variants, where the most frequent mutations are insertions/deletions 50% (22). Despite significant advances in knowledge, 5% to 10% of affected parents with severe deficiencies have no identifiable genetic defect; here the use of next-generation sequencing, correlated with additional investigation on the deleterious/causative role of identified sequence variations, may elucidate novel genetic pathways.
Laboratory Diagnosis
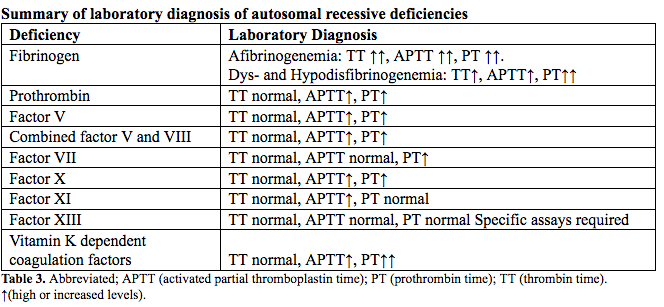
Rare bleeding disorder diagnosis is initially investigated via coagulation screening tests including the APTT (activated partial thromboplastin time) and PT (prothrombin time) (23). A prolonged APTT with normal PT suggests FXI deficiency after exclusion of FVIII, FIX, and FXII deficiencies. The reverse patten is typical of FVII deficiency, whereas the prolongation of both tests directs further analysis toward deficiencies of combined FV and FVIII, FX, FV, prothrombin, or fibrinogen. All coagulation tests depending on the formation of fibrin as the end point are necessary to evaluate fibrinogen deficiency; hence beside the PT and APTT, the TT (thrombin time) is performed. Abnormal screening coagulation tests are followed by mixing studies (50:50) to exclude an inhibitor. When mixing studies correct, specific factor assays are performed to identify the deficiency. Factor antigenic assays are essential for diagnosis of qualitative deficiencies of fibrinogen or FII to appropriately classify and treat patients with dysfibrinogenemia and dysprothrombonemia, both associated with an increased thrombotic risk.
The screening clotting evaluation tests (PT, APTT, fibrinogen, platelet count, and bleeding time) are normal in FXIII deficiency; diagnosis is established via specific assays. Increased clot solubility in 5 M urea, dilute monochloroacetic, or acetic acid is not quantitative or standardized and only detects activity levels <5% resulting in underdiagnosis. Therefore, FXII activity should be quantitatively measured via ammonia release during the transglutaminase reaction or incorporation or radioactive amines into proteins (24). Here plasma blanking is required to avoid the FXIIIa-independent ammonia release that could lead to incorrect result in the low-activity range <5 to 10% (25). If FXIII activity is decreased, the deficiency subtype, FXIII-A or FXIII-B, is determined with an immunological FXIII antigen assay to assure appropriate classification and treatment (26).
Ehlers-Danlos Syndrome
Ehlers-Danlos syndrome (EDS) is a connective tissue disorder with an overall prevalence of 1 in 5,000 (28). It is comprised of 13 clinically heterogeneous conditions characterized by joint hypermobility, skin extensibility, and tissue fragility. Hypermobile subtype (type III) and other hypermobile spectrum disorders such as classic (type I and type II) EDS and vascular (type IV) EDS, are most common, with hypermobile EDS incidence likely greater than 1 in 500 (28). This affects adolescents with heavy menstrual bleeding and other bleeding symptoms and impair normal hemostasis. EDS is more often associated with peripartum complications and increases morbidity and mortality. Vascular (type IV) EDS in particular is associated with uterine rupture, as well as increased risk for preterm delivery, postpartum hemorrhage, and third- or fourth-degree lacerations. Together with inherited coagulation factor disorders, there is an urgent need for early recognition, diagnosis, and intervention in patients with EDS. Because many of these individuals may present with heavy menstrual bleeding, increased screening and referral for confirmation of a hypermobile spectrum disorder diagnosis can potentially improve outcomes over their lifespans.
Developing a National Prevention Campaign
Working form the key findings of the national survey described here and extant knowledge in the field, Women's Health and Education Center (WHEC) with its partners promotes 5 key messages. We believe it would be necessary to in an effective campaign to prevent complications of bleeding disorders. General guidelines for fetal management includes pregnancy counseling, the option of preimplantation genetic testing for hemophilia and other rare bleeding disorders, and consideration of delivery of potentially affected male neonates with hemophilia by cesarean delivery to reduce the risk of neonatal intracranial hemorrhage. In addition, delivery of possibly affected neonates should occur in a facility where there is newborn intensive care and pediatric hemostasis expertise. For patients with other inherited bleeding disorders, unless a severely affected neonate is anticipated, mode of delivery should be dictated by obstetric indications (27). Nonetheless, invasive procedures such as fetal scalp clip or operative vaginal delivery should be avoided, if possible, in any fetus potentially affected with a bleeding disorder.
Do the 5!
- Annual HTC (hemophilia treatment centers) comprehensive checkup;
- Hepatitis A and B vaccination;
- Early & adequate treatment of bleeding episodes;
- Regular physical exercise to protect joints;
- Routine monitoring for blood-borne infections.
It is important to take into account the sensitivities, feelings, fears, and other issues associated with the condition. Equally important is consideration of normative developmental or life stage issues for the target population, particularly when interventions are being targeted toward young people with chronic conditions.
Summary
New assays available for monitoring patients, new therapy, both recombinant and plasma derived, is now available. Registries and clinical trials have demonstrated decrease bleeding and improved outcomes when patients are treated with these agents. Expanding international registries have initiated to correlate genotype and bleeding phenotype in conjunction with global assays. Ongoing research continues to expand our understanding of pathophysiology or rare clotting factor deficiencies. This work complements medical practice to incorporate early diagnosis and new treatment options for patients, resulting in safer and less sensitizing regimens and much improved clinical outcomes.
Suggested Reading
- Managing von Willebrand Disease (VWD) in Women;
http://www.womenshealthsection.com/content/gyn/gyn036.php3 - Vitamin K Deficiency Bleeding
http://www.womenshealthsection.com/content/obsnc/obsnc014.php3 - Overview of Blood Coagulation System;
http://www.womenshealthsection.com/content/obsnc/obsnc015.php3 - • Hemophilia: A Comprehensive Review;
http://www.womenshealthsection.com/content/obsnc/obsnc016.php3
References
- Hsieh L, Nugent Diane. Rare factor deficiencies. Current Opinion in Hematology 2012;19(5):380-384
- Peyvandi F, Palla R, Menegatti M, et al. European registry of rare bleeding disorders. Hematology Education: the education program for the annual congress of the European Hematology Association, 2010, Vol 4 (pg. 63-68). Available online @ href=https://ehaweb.org/
- James AH. More than menorrhagia: a review of the obstetric and gynecological manifestation of bleeding disorders. Haemophilia 2005;11(4):295-307
- Peyvandi F, Palla R, Menegatti M, et al. European Network of Rare Bleeding Disorders Group Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thormb Haemost 2012;10(4):615-621
- de Moerloose P, Casini A, Neerman-Arbez M. Congenital fibrinogen disorders: an update. Semin Thromb Hemost 2013;39(6):585-595
- Sorensen B, Bevan D. A critical evaluation of cryoprecipitate for replacement of fibrinogen. Br J Haematol 2010;149(6):834-843
- Lancellotti S, Basso M, De Cristofaro R. Congenital prothrombin deficiency: and update. Semin Thromb Hemost 2013;39(6):596-606
- Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders: diagnosis and treatment. Blood 2015;125(13):2052-2061
- Thalji N, Camire RM. Parahemophilia: new insights into factor V deficiency. Semin Thromb Hemost 2013;39(6):607-612
- Zheng C, Zhang B. Combined deficiency of coagulation factors V and VIII: an update. Semin Thromb Hemost 2013;39(6):613-620
- Mariani G, Bernardi F. Factor VII deficiency. Semin Thromb Hemost 2009;35(4):400-406
- Menegatti M, Peyvandi F. Factor X deficiency. Semin Thromb Hemost 2009;35(4):407-415
- Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost 2013;39(6):621-631
- Kravtsov DV, Wu W, Meijers JC et al. Dominant factor XI deficiency caused by mutations in factor XI catalytic domain. Blood 2004;104(1):128-134
- Schroeder V, Kohler HP. Factor XIII deficiency: an update. Semin Thromb Hemost 2013;39(6):632-641
- Perez DL, Diamond EL, Castro CM, et al. Factor XIII deficiency related recurrent spontaneous intracerebral hemorrhage: a case and literature review. Clin Neurol Neurosurg 2011;113(2):142-145
- National Institute of Health (NIH). U.S. National Library of Medicine. ClinicalTrails.gov NCT00713648, Evaluation of Recombinant Factor XIII for prevention of bleeding in patients with FXIII inherited deficiency. Available @ https://clinicaltrials.gov/ct2/show/NCT00713648?term=00713648&cntry=US&draw=2&rank=1 last retrieved 20 February 2023
- Brenner B, Kuperman AA, Watzka M, Oldenburg J. Vitamin k-dependent coagulation factors deficiency. Semin Thromb Hemost 2009;35(4):439-446
- van Geffen M, Menegatti M, Loof A, et al. Retrospective evaluation of bleeding tendency and simultaneous thrombin and plasmin generation in patients with rare bleeding disorders. Haemophilia 2012;18(4):630-638
- Rugeri L, Quelin F, Chatard B, De Mazanrcourt P, Negier C, Dargaud Y. Thrombin generation in patients with FXI deficiency and clinical bleeding risk. Haemophilia 2010;16(5):771-777
- Peyvandi F, Palla R, Menegatti M, Mannucci PM. Introduction: rare bleeding disorders – general aspects of clinical features, diagnosis, and management. Semin Thromb Hemost 2009;35(4):349-355
- Peyvandi F, Kunicki T, Lillicrap D. Genetic sequence analysis of inherited bleeding diseases. Blood 2013;122(20):3423-3431
- Kitchen S, McCraw A, Echenagucia M. Diagnosis of hemophilia and other bleeding disorders. A laboratory manual, 2nd edition. Available @ https://www.wfh.org/en/home Last accessed 2 March 2023
- Muszbek L, Bagoly Z, Cairo A, Peyvandi F. Novel aspects of factor XIII deficiency. Curr Opin Hematol 2011;18(5):366-372
- Ajzner C, Muszbek L. Kinetic spectrophotometric factor XIII activity assays: the subtraction of plasma bank is not omissible. J Thromb Haemost 2004;2(11):2075-2077
- Kohler HP, Ichinose A, Seitz R, Ariens RA, Muszbek L. Factor XIII and Fibrinogen SSC Subcommittee Of The ISTH Diagnosis and Classification of factor XIII deficiencies. J Thromb Haemost 2011;9(7):1404-1406
- Pacheco LD, Saade GR, James AH. Von Willebrand disease, hemophilia, and other inherited bleeding disorders in pregnancy. Obstet Gynecol 2023;141:493-504
- Wright TS, Cygan PH. Closing the diagnostic gap in adolescents and young adults women with bleeding disorders. Obstet Gynecol 2023;142:251-256
Опубликован: 18 August 2023
Dedicated to Women's and Children's Well-being and Health Care Worldwide
www.womenshealthsection.com