


Гемофилия: всесторонний обзор
Бюллетень WHEC Практика и клинической управления для медицинских работников. Образование гранта, предоставленного здоровья женщин и образовательный центр (WHEC).
Hemophilia, which means love (philia) of blood (hemo), is the most common severe hereditary hemorrhagic disorder. Hemophilia has often been called “the disease of the kings,” as is often described in the descent of Queen Victoria of England. The earliest description of ancient history dates from second century AD in Babylonian Talmud about a woman who had lost her first two sons from circumcision. The earliest description in modern history was documented by the American physician Dr. John Conrad Otto. He described and inherited bleeding disorder in several families where only males born from unaffected mothers are affected. He then called them the “bleeders.” Hemophilia, as a word, was first documented by Johann Lukas Schonlein in his dissertation at the University of Zurich, Switzerland. Dr. Nasse was the first to publish the genetic description of hemophilia in Nasse’s Law: which states that hemophilia is transmitted entirely by unaffected females to their sons (1). Hemophilia is the most common of the severe bleeding disorders and if not properly managed since early infancy can lead to chronic disease and lifelong disabilities.
The purpose of this document is to identify the etiology of hemophilia, review the evaluation of hemophilia, outline the treatment and management options available for hemophilia and describe interprofessional team strategies for improving care coordination and communication to advance hemophilia and improve outcomes. The principal aim of care should be to avoid and treat bleeding. The patient should receive treatment in a comprehensive treatment center where interprofessional services are offered at all times to the patients and their families. Hemophilia should be considered in the neonatal period in the case of unusual bleeding or in case of positive family history.
Etiology
Hemophilia is usually an inherited condition and is caused by the deficiency of clotting factors in the blood. It is always due to a defect or mutation in the gene for the clotting factor. Research has identified over 1,000 mutations in the genes encoding factor VIII and IX, and around 30% are due to spontaneous mutation (2). The encoding genes for factors VIII and IX are present in the long arm of chromosome X. Both hemophilia A and B are inherited via an X-linked recessive pattern where 100% of females born from affected fathers will be carriers, and none of the males will be affected. Female carrier mothers have a 50% chance of having affected males and a 50% chance of having carrier females. Females could also be affected if there is a complete inactivation of chromosome X through lionization, partial or complete absence of chromosome X such as Turner Syndrome or if both parents carry the abnormal gene (3).
Epidemiology
Hemophilia is equally distributed among all ethnic groups worldwide. The estimated frequency of hemophilia is around 1 in 10,000 live births, and the number of people worldwide living with hemophilia is about 400,000 (4). Hemophilia A is more prevalent (80% to 85% of the total hemophilic population) than hemophilia B. it presents in 1 in 5,000 live births, whereas hemophilia B presents in 1 in 30,000 live births. Due to its X-linked inheritance pattern, geographical areas with a higher frequency of consanguineous marriages like Egypt have higher prevalence of the disease. Hemophilia C occurs in 1 of every 100,000 people. However, Ashkenazi Jews have a higher incidence of factor XI deficiency, which is around 8% (5). With new advances in early diagnosis and treatment therapies, affected individuals should expect a normal life expectancy.
Pathophysiology
The process of blood clot formation involves the activation of two pathways – the extrinsic or tissue factor (TF) pathway and the intrinsic or the contact pathway. Both pathways consist of a series of cascade enzyme activation events that lead to the formation and stabilization of a blood clot by crosslinking of fibrin monomers and activation of platelets. The extrinsic pathway gets triggered by disruption of the endothelium and exposure of TF in the subendothelium. Tissue factor then binds activated factor VIIa forming a complex, which activates factors IX and X into IXa and Xa, respectively. The intrinsic pathway becomes activated when factor XII, prekallikrein (PK), and high-molecular-weight kininogen in the blood become exposed to an artificial surface. Factor XII undergoes a conformational change resulting in the small generation of factor XIIa, which activates PK to kallikrein with reciprocal activation of factor XII to XIIa. This resulting generation of factor XIIa activates factor XI to factor Xia, which converts factor IX to factor IXa. Both pathways converge at the production of factor Xa. Factor Xa converts prothrombin (factor II) into thrombin (factor IIa).
Thrombin, in turn, helps release factor VIII from the von Willebrand factor and activates into factor VIIa, activates platelets by exposing phospholipids that bind IXa, and also activates factor XIII into factor XIIIa, which helps to stabilize the clot by cross-linking fibrin monomers. Factor IXa, together with factor VIIa, calcium, phospholipids, form a complex that recruits large quantities of factor X to activate it. In turn, factor Xa together with the prothrombinase complex calcium and phospholipids, help convert prothrombin into thrombin. Thrombin then helps split fibrinogen into fibrin monomers. When factor VIII and factor IX are deficient or dysfunctional, the intrinsic pathway of the coagulation cascade cannot be appropriately activated, thus making the process of clot formation deficient (6).
Types of Hemophilia
Three types of hemophilia exist. These are hemophilia A, hemophilia B and hemophilia C.
- Hemophilia A, the most common form of hemophilia is associated with the deficiency of factor VIII.
- Hemophilia B, also known as Christmas disease, is secondary to deficiency of factor IX.
- Hemophilia C, is only found in 1% of the population and is due to deficiency of factor XI.
Hemophilia can be mild, moderate, or severe depending on the level of clotting factor:
- Normal: factor VIII or IX activity 50 – 150%.
- Mild hemophilia: factor VIII or IX activity 5 – 40%;
- Moderate: factor VIII or IX activity 1 – 5%;
- Severe: factor VIII or IX activity <1%
What are the chances a baby will have hemophilia?
Genes are found on chromosomes. Two of these chromosomes (called X and Y) decide a person’s sex. Females are born with two X chromosomes (XX). Males are born with one X and one Y (XY). The hemophilia gene is carried on the X chromosome. A man with hemophilia passes the hemophilia gene to all of his daughters, but not to his sons. His daughters are called carriers because they carry the hemophilia gene.
A female inherits two copies of the factor VIII or factor IX gene, one from her mother and one from her father. A female can also rarely have hemophilia if she inherits hemophilia alleles from both of her parents or if she inherits one hemophilia allele and her other X chromosomes is missing or does not work properly. Hemophilia may be hidden in a family for many generations if it passes only through females who do not have bleeding symptoms.
When a carrier has a baby, there is a one in two chance that she will pass on the hemophilia gene. If she passes the hemophilia gene to a son, he will have hemophilia. If she passes the hemophilia gene to a daughter, she will be a carrier like her mother. Sometimes a baby is born with hemophilia although his mother is not a carrier. This is because the factor VIII or IX gene changed only in the baby’s body. One in three babies has no family history of hemophilia.
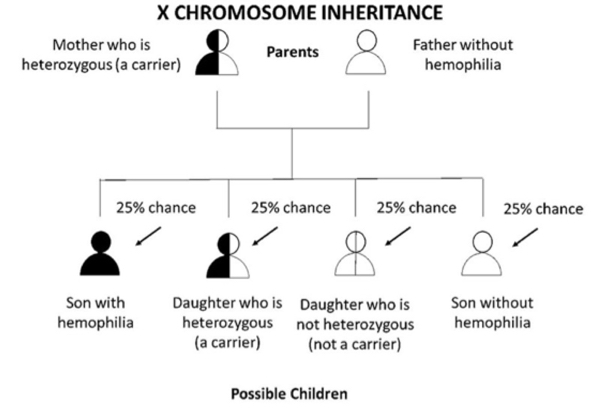
Figure 1 A. If a mother is heterozygous (a carrier) for hemophilia and the father does not have hemophilia, each son has a 1 in 2 (50%) chance of getting his mother’s hemophilia allele and having hemophilia. Each daughter has a 1 in 2 (50%) chance of getting her mother’s hemophilia allele and being heterozygous. Overall, there is a 1 in 4 (25%) chance that the baby will be heterozygous daughter. There is a 1 in 2 (50%) chance the baby (either son or daughter) will not get hemophilia allele at all, and therefore, cannot pass it down to his or her children.
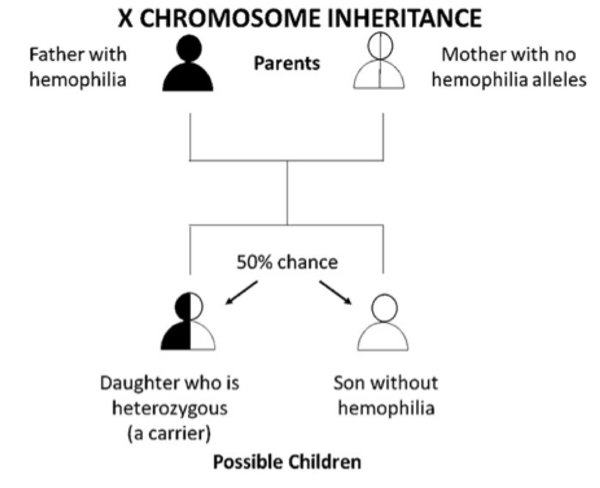
Figure 1 B. If a father who has hemophilia passes his only X chromosome down to all of his daughters, so they will always get his hemophilia allele and be heterozygous (carriers). A father passes down his Y chromosome to his sons; thus, he cannot pass down a hemophilia allele to them. Without the hemophilia allele the sons will not have hemophilia and cannot pass it down to their children. Overall, there is a 1 in 2 (50%) chance that the child will be son who does not have hemophilia and a 1 in 2 (50%) chance that the child will be daughter who is heterozygous (a carrier). This is true if the mother does not have a hemophilia allele herself. That would be very rare, unless the parents are related.
Factor VIII and von Willebrand Factor (VWF): FVIII-VWF Association
A normal hemostatic response to vascular injury requires both factor VIII (FVIII) and von Willebrand factor (VWF). In plasma, VWF and FVIII normally circulate as a non-covalent complex, and each has a critical function in the maintenance of hemostasis. Furthermore, the interaction between VWF and FVIII plays a crucial role in FVIII function, immunogenicity, and clearance, with VWF essentially serving as a chaperone for FVIII. Several novel recombinant FVIII (rFVIII) therapies for hemophilia A have been in clinical development, which aim to increase the half-life of FVIII (~12 hours) and reduce dosing frequency by utilizing bioengineering techniques including PEGylation, Fe fusion, and single-chain design. However, these approaches have achieved only moderate increases in half-life of 1.5- to 2-fold compared with marketed FVIII products. Clearance of PEGylated rFVIII, rVIIIFe, and rVIII-Single Chain is still regulated to a large extent by interaction with VWF. Therefore, the half-life of VWF (~15 hours) appears to be the limiting factor that has confounded attempts to extend the half-life of rFVIII. A greater understanding of the interaction between FVIII and VWF is required to drive novel bioengineering strategies for products that either prolong the survival of VWF or limit VWF-mediated clearance of FVIII
(7).Although it is now well recognized that VWF and FVIII are two separate gene products, the presence of a VWF-FVIII protein complex led to early misunderstanding about relationship. Once VWF was recognized in 2 cell types: megakaryocytes and endothelial cells, processed and then stored in platelet α-granules and endothelial Weibel-Palade bodies, respectively (8).
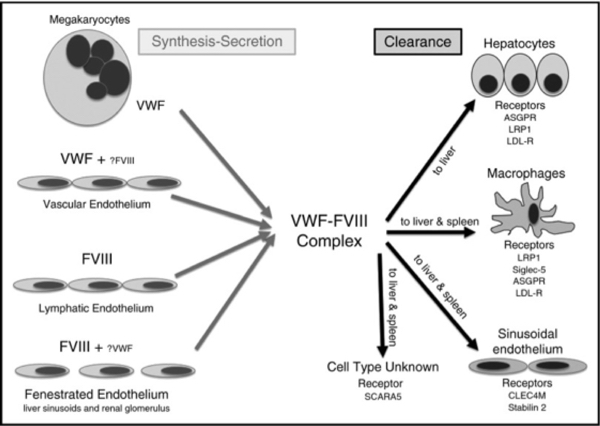
Figure 2. Details of the sites of synthesis and clearance of VWF and FVIII.
Is Hemophilia Lifelong?
A person born with hemophilia will have it for life. The level of factor VIII or IX in his blood usually stays the same throughout his life.
History and Physical
Hemophilia usually presents as bleeding after minor trauma or as a spontaneous bleed. Bleeding symptoms often correlate with the degree of residual factor level, which is useful to classify hemophilia severity further. Patients with greater than 5% to 40% of factor activity of normal (mild hemophilia) often present with bleeding only after significant trauma or surgery. Spontaneous bleeding is uncommon in mild hemophilia. Typically the diagnosis is made incidentally or on routine presurgical laboratory testing. If 1% to 5% factor activity is of normal is present (moderate hemophilia), bleeding usually presents after trauma, injury, dental work, or surgery. In moderate disease, recurrent joint bleed may be present in up to 25% of cases, and the diagnosis usually gets delayed. If factor activity is less than 1% normal (severe hemophilia), bleeding often presents spontaneously. Severe hemophilia usually manifests in the first few months of life, while mild or moderate hemophilia can present later in childhood or adolescence. Recurrent frequent bleeding presents as early as in utero to the lack of transplacental passage of both factor VIII and IX from mother to the fetus.
In cases of severe hemophilia, patients often present with internal bleeding, potentially impacting multiple organs. Joints can become painful, swollen, inflamed, warm, and have a restricted range of motion due to bleeding. The most common affected joints are knees, elbows, ankles, shoulders, wrist, and hips.
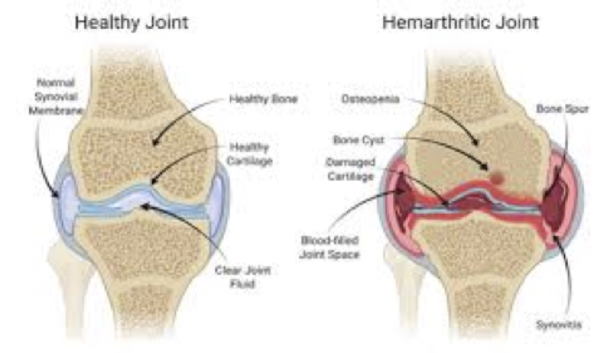
Figure 3. Joint bleed in hemophilia and what happens in joint bleed.
Spontaneous joint bleed incidence typically increases with age reaching up to 60% by 65 years of age (9). Repetitive joint bleeds often lead to hemophilic arthropathies. Usually, hemarthroses become more frequent as physical increases. Brain bleeds, both intracranial and extracranial, are common, and patients can present with falls, confusion, lethargy, meningismus, and coma in severe cases. Intracranial hemorrhage is the earliest and most severe complication in the neonatal period in about 1% to 4% cases (10). Extracranial bleeds such as subgaleal bleed and cephalohematoma can also be part of the initial presentation (11).
What Causes Joint Bleed?
The place where two bone meet is called a joint. The ends of the bones are covered with a smooth surface cartilage. The bones are partly held together by a joint capsule. The joint capsule has a lining called synovium with many capillaries (very small blood vessels). It makes a slippery, oily fluid that helps the joint move easily. If the capillaries in the synovium are injured, they bleed. Often there is no clear reason for the bleed, especially in severe hemophilia, the clotting system stops the bleeding quickly. But in hemophilia, the bleeding continues. This causes the joint to swell and become painful. A person with hemophilia knows when a bleed starts because the joint feels tingly and warm. As blood fills the capsule, the joint swells and becomes painful and hard to move. Without treatment, the pressure from the swelling eventually stops the bleed. Later, special cells clear most of the blood out of the joint.
The most common joint bleeds happen in ankles, knees, and elbows. Bleeds into other joints can also happen, including the toes, shoulders, and hips. Joints of the hands are not usually affected except after the injury. Long-term effects of Joint Bleed: Repeated bleeding into a joint causes the synovium to swell and bleed very easily. Some blood remains in the joint after each bleed. The synovium stops producing the slippery, oily fluid that helps joint move. This damages the smooth cartilage that covers the ends of the bones.
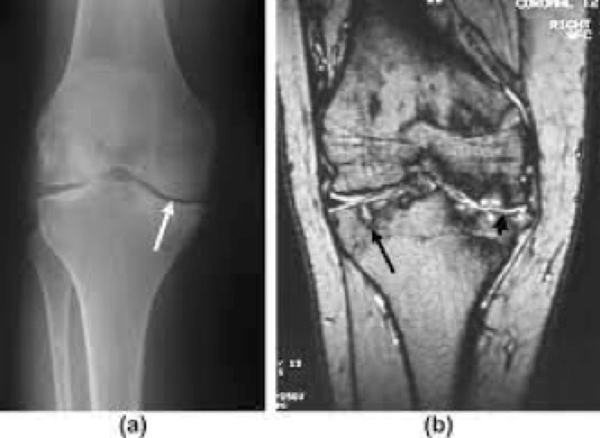
Figure 4. Hemophilic arthritis of knee.
The joint becomes stiff, painful to move, and unstable. It becomes more unstable as muscles around the joint weaken. With time, most of the cartilage breaks down and some bone wears away. Sometimes the joint cannot move at all. The whole process is called hemophilic arthritis.
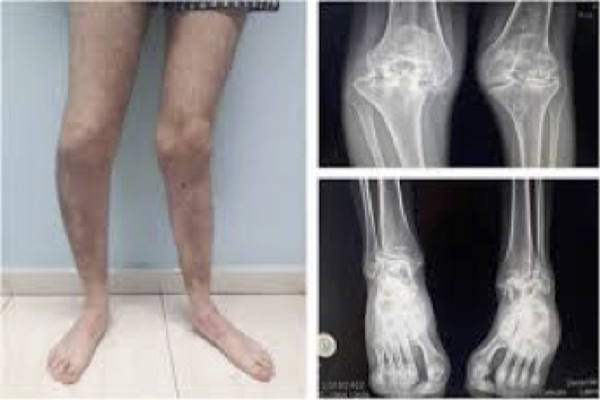
Figure 5. Hemophilic arthropathy. Musculoskeletal disorder.
Muscle Bleed and Long-Term Effects
Muscle bleeds happen when capillaries in the muscle are injured. Sometimes the cause is known, but bleeds can also happen for no clear reason. During a bleed, the muscle feels stiff and painful. The bleed causes swelling that is worm and painful to touch. There may be bruising if the bleed is near the skin. In some of the deeper muscles, the swelling may press on nerves or arteries, causing tingling and numbness.
The muscle tightens up to protect itself. This is called a muscle spasm. As a result, joints that are usually moved by that muscle do not move properly. Muscle bleeds happen in the calf, thigh, and upper arm. Bleeds in psoas muscle (at the front of the hip) and forearm are also common. These bleeds can put pressure on nerves and arteries, causing permanent damage. Bleeds into the muscles of the hand are rare and usually follow an injury.
Hemophilia A
Classical hemophilia, also known as hemophilia A, is a hereditary hemorrhagic disorder resulting from a congenital deficit of factor VIII that manifests as protected and excessive bleeding either spontaneously or secondary to trauma. An X-linked, recessive hemorrhagic trait or gene induces hemophilia A. Its X-linked trait manifests as a congenital absence or decrease in plasma clotting Factor VIII, a pro-coagulation cofactor and robust initiator of thrombin that is essential for the generation of adequate amounts of fibrin to form a platelet-fibrin plug at sites of endothelial disruption. Female hemophilia A gene carriers will transmit the gene to 50% of their male offspring, who will inherit the disorder. Female hemophilia gene carriers do not manifest symptoms of hemophilia A but may have lower than usual quantities of Factor VIII. Male hemophilia patients do not transmit hemophilia to male offspring, but their female offspring will carry the hemophilia gene (12).
Evaluation: Diagnostic evaluation for hemophilia occurs in the setting of known family history, excessive bleeding out of proportion to the traumatic injury, or abnormally activated partial thromboplastin time (aPTT). Normal hemogram and prothrombin time (PT) in the setting of elevated PTT heightens the suspicion of hemophilia and should prompt factor VIII and IX determination. Determining residual plasma concentration of factor VIII represents the keystone of diagnosis, classification, and treatment of hemophilia A as therapy and prognosis will vary depending on factor VIII deficiency. Most hemophilia A patients have a prolonged aPTT; however, normal result does not rule out mild hemophilia.
Hemorrhage severity in hemophilia A correlates scarcity of factor VIII. Factor VIII concentration, expressed in international units (IU); 1 IU is the concentration of factor VIII in 1 mL of pooled plasma or percentages of normal pooled plasma with normal levels ranging between 50% to 150%. Severe hemophilia A will have no measurable factor VIII, less than 0.01 IU/mL (2% to 5%) or 0.06 IU/mL to 0.40 IU/mL (6% to 40%) respectively, will bleed excessively after relatively insignificant trauma (13).
Treatment / Management
Administration of recombinant factor VIII replacement for the treatment of acute bleeding in severe hemophilia A patients should occur promptly with initiation before completion of the patient assessment. Calculation of factor VIII replacement for bleeding in severe hemophilia A is (14):
Dose of factor VIII = percentage desired of factor X bodyweight (kg) X 0.5
For severe, life-threatening hemorrhage, administration factor VIII to achieve a 100% desired factor VIII level; for mild to moderate hemorrhage, administer factor VIII to achieve a 30% to 50% desired factor VIII level. Accounting for the hemophilia A patient’s native factor VIII levels should be factored into factor VIII repletion if known. The combination of effective blood product screening with viral inactivation protocols and recombinant production of Factor VIII have enhanced factor VIII replacement products safety from the viral transmission, such as HIV and hepatitis C.
Desmopressin (DDAVP): Intravenous, subcutaneous, or intranasal DDAVP has utility for the treatment of bleeding in mild to moderate hemophilia A patients by triggering the release of complexes VWF and factor VIII from vascular endothelial cells. Other pharmaceutical adjuvant therapies for hemophilia A-induced bleeding include tranexamic acid, epsilon aminocaproic acid, and management of factor VIII inhibitors.
Periodic, prophylactic Factor VIII concentrates infusions for severe hemophilia A patients have benefit in preventing spontaneous bleeding. The intent of factor VIII prophylaxis aims to modify severe hemophilia to a milder form by keeping the nadir level of factors more than 1% of normal. The World Federation of Hemophilia recommends factor VIII prophylaxis initiation in hemophilic children after their first or second episode of hemarthrosis to prevent joint destruction and preserve musculoskeletal function. Mild and moderate hemophilia A patients receive factor VIII concentrate or desmopressin to prevent hemorrhage in anticipation of trauma or surgery (15).
Hemophilia B
Hemophilia B is an inherited disease, mainly caused by deficiency of factor IX. It mostly affects males, but carrier families may show some signs of bleeding. It has an X-linked recessive inherited mode of inheritance, but some acquired form has also been reported due to development of autoantibodies toward factor IX. Hemophilia B is also known as Christmas disease, is the second most common type of hemophilia. The disease was named after Stephen Christmas, who was the first person diagnosed with the condition in 1952. The disorder was ubiquitous in the royal families of Spain, Germany, and Russia. One of the famous families with this condition was that of Queen Victoria of England; thus, it is known as the “Royal Disease.” It is a hereditary hemorrhagic disorder resulting from congenital deficit or scarcity of factor IX, which manifests either spontaneously or after traumatic events (16).
The prevalence of hemophilia B is 1 in 40,000 live males which is about 15% of cases of hemophilia, while that of hemophilia A is 1 in 5,000 live males which is about 85% of cases of hemophilia. Factor IX is synthesized by the hepatocytes and considered to be part of the intrinsic pathway, and its deficiency will result in the defective coagulation cascade and insufficient fibrin mesh formation (17).
History and Physical: Hemophilia B is clinically less severe than hemophilia A (18). In the neonatal period, the presentation usually start when the infant starts to walking or crawling, or after circumcision or getting intramuscular vaccinations. The hallmark clinical presentation of hemophilia B is joint involvement (hemarthroses), which typically presents in severe disease. The most common joints affected are knees, elbows, ankles, shoulders, wrist and hips.
Evaluation: Patients with hemophilia B will present with decreased factor IX level. The patient could also present with prolonged aPTT, which indicate an intrinsic pathway disruption, but a normal aPTT and a normal PT does not exclude hemophilia. Complete blood count could show normal or low hemoglobin levels with normal platelet levels. After blood test suggest hemophilia, then a coagulation factor VIII and IX should be the next step. Factor IX is present in plasma in a concentration of 4-5 μg/mL. Thus the disease is categorized according to scarcity of factor IX (19).
Mild hemophilia B: if 5% to 50% of factor IX is present. Moderate hemophilia B: If only 1% to 5% of the factor IX activity of normal is present. Severe hemophilia B: If less than 1% of factor IX is present.
Molecular genotyping should then be offered to confirm the diagnosis. In patients with an established diagnosis of hemophilia B, periodic laboratory evaluation include screening for the presence of factor IX antibodies and testing for transfusion-related infections such as hepatitis and HIV.
Laboratory features: Normal platelet count; prolonged activated partial thromboplastin time (aPTT) in severe and moderate hemophilia B. Normal or mildly prolonged aPTT in mild hemophilia B. Normal prothrombin time (PT). Identification of a hemizygous pathogenic variant in F9 by molecular genetic testing can help predict the clinical phenotype and allow family studies.
Treatment
Management of acute bleeding of hemophilia B patients:
- Replacement of factor IX remains the mainstay of treatment for hemophilia B patients. The dose is calculated according to the desired percentage of the factor with a goal of 30% in patients with mild hemorrhages, 50% in patients with severe bleed after trauma or prophylaxis of major dental surgery or major surgery, and 80% to 100% in patients with life-threatening conditions. It is calculated with this formula (20): Initial dose = body weight (kg) X desired factor IX increase (% or IU/dL) X reciprocal of observed recovery (IU/dL per IU/kg). If there is no available factor IX then prothrombin complex concentrate can be given at a dose of IU/kg.
- Monoclonal antibodies (e.g., rituximab), tranexamic acid, and epsilon aminocaproic acid are anti-fibrinolytic agents that could be used in cases of mucosal bleeds and dental extractions in patients with hemophilia.
Prophylaxis
The main goal of factor IX prophylaxis therapy is to improve the quality of life through reducing hemarthrosis episodes, its development into hemophilic arthropathy, and the need for surgeries and minimizing the episodes of cerebral and muscle bleeds. The goal of dosing has been to maintain factor IX level above 1 to 2%, mainly converting the patients from a severe to a moderate hemophilia phenotype. Factor IX dosing, either 25 or 40 units/kg two times per week (Utrecht protocol). Longer lasting products allow for once per week or once every two-week dosing (21).
Future Treatment Considerations
The novel treatments like genetic therapy, through introducing exogenous DNA into a person’s cells that can be used to produce a missing protein (22). Also cellular therapy, through injecting intact cells into the patient rather than the manipulation of coagulation factor genes have been under testing, and monoclonal antibodies like concizumab which is an antibody directed against the tissue factor pathway inhibitor (TFPI), which inhibits the coagulation cascade by blocking the function of factor Xa. Blocking the inhibitor allows the generation of factor Xa and in turn, thrombin, even in the absence of factors VIII and IX.
Inhibitors Development
A significant complication in patients getting a replacement of factor IX is the development of antibodies (IgG) that block the activity of the replaced factor. These inhibitory antibodies develop in response to exogenous factors. They occur in approximately 3 to 5% with severe hemophilia B. Inhibitors are much less common in patients with mild to moderate disease, mainly because the infused factor does not get recognized as foreign protein in these individuals. Inhibitors complicate bleeding episodes because they decrease responsiveness to factor infusions (23). The presence of inhibitors should be suspected if bleeding does not stop after clotting factors replacement in a previously responsive patient. Also, anaphylactic reactions can occur with factor IX inhibitors. These inhibitors are managed by:
- Recombinant activated FVIIa administration: these contain an activated form of a downstream clotting factor in the coagulation cascade.
- Plasmapheresis: it may be useful in patients with a high titer inhibitor to acutely lower the inhibitor titer and allow transient use of replacement factor. That intervention is reserved for an individual with life-threatening or limb-threatening bleeding.
- High-dose factor infusion: a final option for an individual with bleeding, and an inhibitor is to provide high-dose factor infusion.
Hemophilia C
The incidence of hemophilia C is 1 in 100,000, which is about 1% of hemophilia cases. It is due to factor XI deficiency, which causes abnormal bleeding. This condition is classified as either partial or severe based on the degree of deficiency of the factor XI protein. However, regardless of the severity of the protein deficiency, most affected individuals have relatively mild bleeding problems, and some people with this disorder have few if any symptoms. The most common feature of factor XI deficiency is prolonged bleeding after trauma or surgery, especially involving the inside of mouth and nose or the urinary tract. If the bleeding is left untreated after surgery, solid swelling consisting of congealed blood (hematomas) can develop in the surgical area.
The severe deficiency disorder is much more common in people with central and eastern European (Ashkenazi) Jewish ancestry, occurring in about 1 in 450 individuals in that population. Researchers suggest that the actual prevalence of factor XI deficiency may be higher than reported, because mild cases of the disorder often do not come to medical attention.
Causses: Most cases of factor XI deficiency are caused by mutations in the F11 gene, which provides instructions for making the factor XI protein. This protein plays a role in the coagulation cascade, in response to injury. Mutations in the F11 gene result in a shortage (deficiency) of functional factor XI. Some cases of factor XI deficiency are not caused by F11 gene mutations. In these cases, the condition is called acquired factor XI deficiency. It can be caused by other disorders such as conditions in which the immune system malfunctions and attacks the factor XI protein. Because factor XI is made primarily by cells in liver, acquired factor XI deficiency can also occur as the result of severe liver disease or receiving a transplanted liver from an affected individual. In addition, approximately 25% of people with another disorder called Noonan syndrome have factor XI deficiency.
Other Names for This Condition: F11 deficiency; Factor 11 deficiency; Hemophilia C; Haemophilia C; Plasma thromboplastin antecedent deficiency; PTA deficiency; Rosenthal factor deficiency; Rosenthal syndrome; and Rosenthal’s disease.
Laboratory studies: aPTT should be measured. In patients with severe factor XI deficiency, the aPTT value will be more than 2 standard deviations above the normal mean; in heterozygotes, the aPTT may be slightly prolonged or within normal range (24). An FXI assay may help confirm the diagnosis, although levels can be in the normal range. Homozygotes and compound heterozygotes will have an FXI level of less than 15%. The expected FXI level in heterozygotes is 25 – 70%. Researchers are exploring the use of novel laboratory tests, such as thrombin generation assays and clot stability assays, to predict bleeding risk in patients with FXI deficiency (25). Combining the aPTT with the rate of clot formation and area under the curve in fibrinolysis assays identifies most FXI-deficient patients with a bleeding tendency.
Treatment / Management
Patients with factor XI deficiency do not need treatment or prophylaxis for routine functions or activities, but may need treatment for dental extractions, surgery and childbirth (26). Treatment of FXI deficiency is determined by patient factors and clinical circumstances. Fresh Frozen Plasma (FFP) has been the most available source of FXI. The recovery of FXI function from plasma is excellent, and the half-life is 40 to 80 hours. FXI concentrates are produced in the United Kingdom and France, but are not available in the United States. Dental extractions have been performed safely in severely FXI-deficient patients with the use of antifibrinolytic therapy alone. Tranexamic acid, 1 g four times daily, was started 12 hours before the procedure and continued for 7 days afterward. Antifibrinolytic therapy has also been used in the treatment of women with FXI deficiency and menorrhagia (27).
Pregnant women with need FFP if cesarean delivery is planned. Peripartum treatment of women with FXI deficiency is controversial. The use of desmopressin, a vasopressin analog, used for patients with factor VIII deficiency, VWF deficiency, and platelet function abnormalities, has been tried in a handful of patients with FXI deficiency. The true benefit of this treatment is unclear, and it is not recommended for major surgical procedures.
Treatment of patients with acquired antibodies to FXI has not been standardized because of the infrequency of this occurrence. Successful treatment has been reported during invasive procedures with the use of FFP, prothrombin complex concentrates, and rFVIIa. Reports also exist of patients with inhibitors who have no spontaneous bleeds. Unless needed for another medical indication, aspirin products should be avoided by patients with FXI deficiency. Immunization with hepatitis A virus and hepatitis B virus vaccines is recommended prior to planned surgery and plasma product replacement. Consultation with hematologist is recommended.
Acquired Hemophilia (AHA)
The overall incidence of acquired hemophilia is 1 in 1.5 per million per year (28). However, the incidence varies with age from 0.045 per million per year in children younger than 16 years of age to 14.7 per million per year in adults older than 85 years. hence, the disease is more common in adults than in children. There are reports that 80% of the patients are older than 65 years. the medial age ranges from 73.9 years to 78 years. There has been no evidence of any genetic pattern and is overall equal in males and females.
Acquired hemophilia is an uncommon but severe bleeding disorder. It is caused by the development of autoantibodies directed against one of the antihemophilic factors, most frequently factor VIII (FVIII). It is commonly known as “acquired hemophilia A” (AHA) (29). Various factors that can predispose to the development of AHA may include the following:
- Immunological Disorders (17 to 18%). Associated with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Sjogren syndrome, connective tissue diseases, autoimmune thyroiditis, Grave disease, antiphospholipid syndrome, multiple sclerosis, temporal arteritis, myasthenia gravis, Goodpasture syndrome, and other autoimmune diseases (30).
- Obstetrical Causes. Postpartum-state (one of the common causes of acquired hemophilia about 8.4% of total incidence). Any episode of abnormal bleeding in postpartum period (up to 12 months after delivery) should raise the suspicion for AHA.
- Hematological and Oncological Causes. Diagnosis of cancer, precancerous states, solid tumors (lung, prostate, pancreas, breast), hematological neoplasia, chronic lymphocytic malignancies, non-Hodgkin lymphoma, monoclonal gammopathy of undermined significance (MGUS) and polymyalgia rheumatica (31).
- Dermatologic Disorders. Psoriasis, pemphigus, and epidermolysis bullosa.
- Pharmacological Causes. Beta-lactam antibiotics, penicillin, non-beta-lactam antibiotics, interferons, clopidogrel, non-steroidal anti-inflammatory drugs (NSAIDS), amiodarone, rivastigmine, sunitinib, heparin, phenytoin, chloramphenicol, methyldopa, and fludarabine.
- Infectious Diseases. Acute hepatitis B and C infections and others.
- Transplant Related Disorders. Chronic graft versus host disease.
- Gastroenterological Diseases. Inflammatory bowel disease.
- Pulmonary Diseases. Asthma, chronic obstructive pulmonary disease (COPD).
- Transfusion of blood products.
Evaluation: When the history and physical examination raise suspicion, the diagnosis of AHA should be confirmed by a complete blood count and a coagulation profile. The complete blood tests reveals a normal platelet count, whereas the coagulation profile shows an isolated prolonged aPTT by 2 to 3 times its normal value. Prolonged aPTT can be attributed to the deficiency of multiple factors of the intrinsic pathway, including FVIII, FIX, FXI, FXII, as well as the presence of antibodies against these factors. To rule out either of the causes of the functional deficiency of the factors mixing studies are helpful. The antibody titers are described in the Bethesda unit (BU), which represents the amount of inhibitor that leads to 50% residual FVIII:C activity. The Nijmegen modification to the Bethesda method has helped to standardize the acidity as well as the concentration of protein in the testing mixture, which reduces the probability of an artifactual measurement, hence leading to increased specificity of the test (32)
Treatment / Management
The treatment recommendations usually rely on the clinical judgment and expertise of the providers who have treated patients with AHA. The reason being the unavailability of an international consensus for management. The management of AHA, comprises of two-pronged therapy approach in which both modalities go hand in hand. These are: Hemostasis management and Eradication of inhibitor (33).
Hemostasis Management
The requirement for hemostasis management is primarily dependent on the severity of bleeding, as well as its location. It should be noted that the autoantibody titer is not directly correlated to the bleeding severity.
Minor bleeding: In a patient that presents with mild bleeding and is not undergo an emergent invasive procedure, with autoantibody titers of equal to or less than 5 BU (Bethesda Unit), observation is sufficient. These titers have been reported to disappear in 25% of cases within a few months when these were secondary to pregnancy or antimicrobial therapy. In patients with residual factor VIII levels of more than 5% and autoantibody titers less than 2 BU, desmopressin (0.3 – 0.4 mg/kg) has been reported to be helpful with increasing the factor VIII levels.
Major Bleeding: Patients who present with major bleeding (72%) require initiation of appropriate antihemorrhagic treatment in a timely fashion. If the antibody titer is low <5BU, treatment including human factor VIII or desmopressin, may be considered as the first-line therapy. In cases of high titers, first-line treatment is use of bypassing agents. The bypassing agents are named so because of their mechanism of action. They act by bypassing the need for factor VIII in the coagulation pathway in the production of thrombin. These agents include activated prothrombin complex concentrate (aPCC) and recombinant activated factor VII (rFVIIa). The prothrombin complex concentrate, which is plasma-derived and contains exogenously activated vitamin K dependent factors (factors II, VII, IX, X). The recommended dose of aPCC is 100 U/kg per dose or 200 u/kg/day. The half-life of aPCC is around 4 to 7 hours.
In the case of mucosal bleeding, there is supporting evidence for the concomitant use of antifibrinolytic therapy. Once treatment is successful, to prevent bleeding in the future, there are recommendations for prophylactic therapy by using low doses of bypassing agents (34).
Inhibitor Eradication
The treatment of AHA depends on correcting the pathophysiology that arises due to the presence of factor autoantibody. In the presence of the autoantibody, the patient is always at risk of life-threatening bleeding, and the complete eradication of the inhibitor is vital for improving survival in patients. The first-line treatment includes prednisone (1 mg/kg/day). It can be used alone, or cyclophosphamide (50 to 100 mg/day) can be added, although evidence suggests that the combined use is correlated with better patient outcomes. The inhibitor eradication therapy is considered successful with undetectable titers of autoantibody (<0.6 BU) along with normal factor VIII levels (>50%). Indications for second-line immunosuppression include factor VIII levels not increasing, and autoantibody titers are not decreasing at 3 to 5 weeks with appropriate treatment and adequate patient compliance. Rituximab is an anti-CD20 monoclonal antibody successfully applied in the management of various autoimmune conditions and may be a valuable agent in managing acquired hemophilia. It may be considered in cases of intolerance to standard immunosuppressive therapy or resistance (35). 2-chlorodeoxyadenosine has also been shown to be effective in the treatment of some trials.
Differential Diagnosis
Other conditions can also present similarly with bleeding after minor trauma or spontaneous bleeds and require exclusion before confirming the diagnosis of hemophilia. Some of these conditions include von Willebrand disease, scurvy, diseases of platelet dysfunction, deficiency of other coagulation factors like V, VII, X, or fibrinogen, Ehlers-Danlos syndrome, Fabry disease, disseminated intravascular coagulation (DIC) and child abuse.
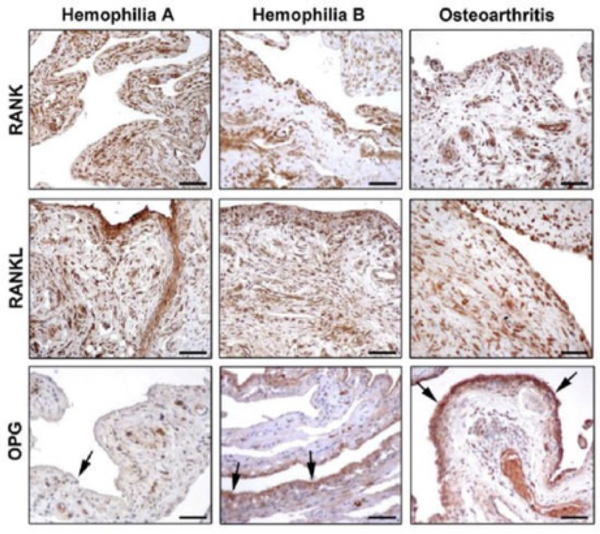
Figure 6. Photomicrographs showing the expression of receptor activator of nuclear factor-kB (RANK), RANK ligand (RANKL), and osteoprotegerin (OPG in synovial tissue from patients with hemophilia A, hemophilia B, and osteoarthritis. Arrows indicate OPG immunostaining in the synovial liming layer.
The diagnosis is usually through clinical features, genetic testing and tissue biopsy (36). Both in hemophilia A and hemophilia B synovium, RANK is strongly expressed in the lining and sublining layers, especially in synoviocytes and vascular endothelium. In osteoarthritis, RANK was less expressed in the lining layer, while a strong immunopositivity was observed in the inflammatory infiltrate of the sublining layer (See Figure 6). The expression of RANL in the lining and sublining layers of hemophilia A and hemophilia B synovium was similar to that observed in osteoarthritis. In conclusion, the reduced number of hemarthrosis, the lower World Federation of Hemophilia and ultrasound scores, and hither osteoprotegerin (OPG) expression in serum and synovial tissue in hemophilia B suggest that hemophilia B is a less severe disease than hemophilia A. OPG reduction seems to play a pivotal role in the progression of arthropathy in hemophilia A.
Prognosis
The life expectancy of people who had severe hemophilia in the 1950s and 1960s before the development of factor concentrates was only 11 years. Most people who had severe hemophilia died in early childhood or adolescence from intracranial bleeds or bleeding inside the vital organs. In the 1970s, lyophilized plasma concentrates of coagulation factors became available, and this improved treatment significantly. Primary prophylaxis began in Sweden before being adopted by other countries, which ended up preventing major bleeding episodes and complications of arthropathies. In 1977, researchers discovered desmopressin. With that, patients were able to get a better, safer, and relatively inexpensive option for treatment, and risks of blood-borne infections from repeated used of plasma-derived products were minimized. Eventually, the advancement in DNA technology allowed the industrial production of recombinant factor VIII and IX.
The widespread availability of replacement therapy to prevent and treat active bleeding, advancement in viral inactivation techniques, management of blood-borne infections through surveillance, and availability of newer treatment options of hepatitis C and HIV treatment have significantly improved the lifestyle of patients with hemophilia. Today, life expectancy of patients is almost the same as the general population in developed countries, provided those patients respond well to the treatment and do not have other health conditions. But in developing countries, where healthcare access and treatment resources are scarce, the mortality rate remains almost twice that of the general population (37).
Hemophilia Care in Pediatric Age
Prophylactic treatment that is started early with clotting-factor concentrates has been shown to prevent hemophilic arthropathy and is therefore, the gold standard of care for hemophilia A and B in most countries with adequate resources. Central venous access catheters and arteriovenous fistulas play an important role in the management of hemophilia children requiring repeated and/or urgent administration of coagulation factor concentrates. During childhood and adolescence, personalized treatment strategies that suit the patient and his lifestyle are essential to ensure optimal outcomes.
Vaccinations: A child with hemophilia can and should be vaccinated like other children. While most vaccines can be given subcutaneously, some are necessary to give intramuscular. The World Federation Hemophilia (WFH) guidelines suggest that, if intramuscular injection is necessary, due to some vaccines having only intramuscular route as suggested route of administration, it is best done soon after a dose of factor replacement therapy, an ice pack should be placed over the injection site 5 minutes before injection, a small gauge needle (23 gauge) should be used, and pressure should be applied on an injection site for 5 minutes after injection (38). As a common trend and as a hypothetical measure to avoid an immune system challenge and inhibitor formation, a vaccination should NOT be given on the same day as factor concentrate infusion. On the other hand, the possible role of vaccinations as “danger signal” for inhibitor development has been described. However, recent data showed that vaccination does not pose any increased risk for inhibitor development.
Product choice: In developed countries, most children with hemophilia are treated with recombinant FVIII/FIX, mainly due to their perceived higher safety. Newer Extended Half-life Recombinant Agents, also known as enhanced half-life (EHL) clotting factors, may help in the future to reduce the frequency of prophylaxis administration.
Regimen Choice and When to Start: The goal of treatment of a child with hemophilia is to enable the patient and their families to manage illness as independently as possible and therefore lead more normal, healthy lives. To date, the most refined regimen involves primary prophylaxis started before the onset of joint bleeding or other serious bleeds, 12 – 18 months of age (38), or earlier as in some centers who start before one year of age. Although treatment should be individualized, taking into account the patient’s age, venous access, bleeding phenotype, susceptibility to joint arthropathy, and availability of clotting factors, the protocol more commonly used for prophylaxis is as follows: in most cases, an early therapeutic approach is initiated by giving approximately 30 – 50 IU/kg once or twice a week, with the aim of increasing the frequency of administration as soon as possible, until reaching full-scale primary prophylaxis. In hemophilia A, FVIII is administered at dosage of 20-40 IU/kg/day every second day or three times weekly; in hemophilia B, FIX is administered at dosage of 20 – 40 IU/kg/day every third day or two times weekly (38).
Inhibitors: The development of inhibitors, which occur in 30 – 50% of previously untreated children with severe hemophilia A and around 2 – 5% of patients with severe hemophilia B infused with the available commercial product, remains the major complication of therapy in hemophilia. Inhibitors are alloantibodies directed against the replacement FVIII or FIX that typically neutralize the activity of the factor. The main goal in patients with inhibitors is to eradicate the antibody; thus, immune tolerance induction (ITI) with high daily doses of FVIII has become the standard therapy; however, no standard dosing schedule has been defined. Early detection of an inhibitor is crucial to minimize anamnesis, and if the inhibitor does not rise about 10 BU/mL, allow ITI to be started without delay. In the case of a high-titer inhibitor >10 BU/mL, it is advisable to assume a “wait and see” strategy initially, monitoring the inhibitor titer at least monthly and ideally start ITI one the liter is <10BU/mL or earlier in case of clinical need, bleeding phenotype for example.
Venous Access: One of the major issues and difficulties in the treatment of hemophilia in children is the venous access. Easy venous access is a prerequisite for treatment of hemophilic patients with factor concentrates, whether this prophylaxis, on-demand or an ITI regimen. The alternative to a peripheral vein in the case of on demand or prophylactic regimen, and a necessity in case of ITI, is a central venous access device (CVAD) or an arteriovenous fistula (AVF) (39). An implantable CVAD such as a Port-a-cath carries a reduced infectious risk compared to a peripherally inserted external non-tunneled CVAD such as peripherally inserted central catheter (PICC) or an external tunneled CVAD like, for example, Hickman-Broviac devices. These catheters carry, furthermore, a thrombotic risk, rare in hemophilia but not absent.
In 2004, and international consensus conference among hemophilia experts stated that, whenever possible, peripheral veins remain the route of choice, and the use of central venous access devices should be limited to cases of clear need in patients with caregivers able to exercise diligence in CVAD care and should continue no longer than necessary. On the other hand, the good outcome obtained with AVF leads to diversely considering this possibility, but the AVF creation has an anatomical age limit based on the size of the brachial artery and the identification of a suitable vascular site by the vascular surgeon, who should be an expert in operating on small caliber vessels (39).
Dental Care: Hemophilic children should be educated to a correct oral hygiene, targeted by brushing twice daily with a medium texture bristles toothbrush and age-adapted fluoride toothpaste, and using interdental cleaning aids, such as floss, tape, and interdental brushes to prevent the formation of dental caries and periodontal disease. Where water does not have a fluoride content of at least 1 ppm, fluoride toothpaste as well as additional fluoride supplements should be recommended. There are four therapeutic management options depending on the type of hemophilia, for dental extractions and procedures:
- Coagulation factor replacement therapy;
- Release of endogenous factor stores using desmopressin (DDAVP);
- Improving clot stability by antifibrinolytic drugs, for example tranexamic acid, to reduce the need for replacement therapy;
- Local hemostatic measures (such as suturing, and local measures, such as the use of oxidized cellulose).
It is advisable that complicated dental procedures, such as dental extraction or surgical procedures carried out within the oral cavity, should be performed in a Hemophilia Treatment Center.
Joint Evaluation: In the frame of a comprehensive care approach, growing attention has been given to the periodic assessment of the joint status in hemophilia patients, with the aim to identify early joint damage and to prevent the development of a clinically overt arthropathy. Besides clinical examinations, X-ray and magnetic resonance imaging (MRI) are currently used to evaluate the joint status and to monitor the disease progression. Ultrasound is also proposed as part of the routine clinical examination to monitor synovial hypertrophy, by hemophilia experts.
From Day-Care to School: finding an out-of-home child care may seem doubly challenging for a parent of a child with bleeding disorder, but day care or other activity groups can provide stimulation that the child needs, avoiding the risk of overprotection. The school staff and students should be informed that a child has hemophilia, preferably by the parents. Education of school personnel regarding suitable activities for the child and immediate care in case of bleed is recommended. If adequate prophylaxis is given, no additional resources are required for medical reasons.
Sports and Hemophilia in Childhood: Physical exercise is one of the basic foundations in the treatment of hemophilia. Specifically, a child with hemophilia would benefit from exercise and sport, both because a good muscle tone can decrease the frequency of bleeds, joint problems and loss of bone mineral density, and because it can contribute to improving their quality of life (40). Furthermore, acute exercise sessions increment the levels of Factor VIII, subsequently improving coagulation in mild patients. Non-contact sports (such as swimming, cycling, and walking) should always be encouraged, high contact sport (soccer, rugby, boxing) or high-velocity activities (skiing, motocross) are best avoided unless the individual is on good prophylaxis to cover such activities. Efforts should be made to maximize safety for patients with hemophilia.
Adolescence: Persons with hemophilia, living with their condition from infancy, require attention from biopsychosocial approach. They may benefit greatly from having professional help to achieve the best quality of life possible setting up tailored objectives throughout the patient’s life, including disease control, addressing the particular difficulties, and achieving optimal empowerment. This becomes even more critical in prepuberty and adolescence, as these periods are considered at risk from a clinical-biological point of view (such as overweight) and a psychological point of view (such as psychosexual and psychosocial regression).
Psychological Issues: Hemophilia dose not predispose to any mental illness, but the person with hemophilia and his environment may greatly benefit from having professionals help them manage to adapt to the disease, cope with the experience of suffering and overcome the difficulties caused by chronicity, achieving the best quality of life (QoL) possible. Psychosocial factors affecting QoL include coping, social support and locus of control that may influence both as resources and stress factors (41).
Gene Therapy for Hemophilia
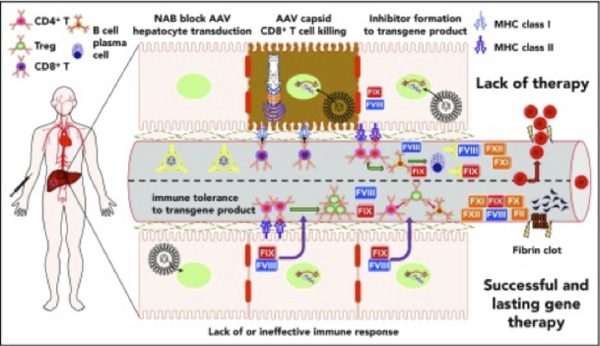
Figure 7. Summary of immunological challenges and successes of liver-directed AAV gene therapy for hemophilia in humans. Details available @ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6356985/figure/F1/
Adeno-associated virus (AAV)- mediated gene transfer has successfully raised, and in some cases transiently normalized, FVIII or FIX activity levels in adults with severe hemophilia. Raising FVII/IX levels, particularly greater than ~15IU/dL (mild deficiency), corresponds to a marked decrease in spontaneous and provoked bleeding, dramatic reduction in factor concentrate use, and improved quality of life. Limited understanding of innate and adaptive immune system responses and hepatocyte transgene expression and stress responses to AAV-medicated gene transfer contribute to the variability in initial and long-term factor protein expression. Lentiviral (LV) and CRISPR/Cas-9 gene therapy approaches may further bolster the range of eligible participants and improve transgene expression and durability (41).
Summary
Defects in the binding of factor VIII and von Willebrand factor, characterizing type 2N or “Normandy type” von Willebrand disease, will manifest like hemophilia-induced bleeding, but unlike hemophilia, inherited manifestation occurs in an autosomal pattern, thereby also affecting female patients. Establishing the diagnosis of type 2N von Willebrand disease utilizes molecular genetic testing. Many severe hemophilia A patients, nearly 30%, develop alloantibodies against administered factor VIII. Inhibitor development represents a major complication and challenges in hemophilia treatment because these alloantibodies inactivate the procoagulant effect of infused factor VIII, thereby inhibiting the response to factor VIII replacement.
Hemophilia management is best undertaken by an interprofessional team, that includes hematology nurses. Once the diagnosis is confirmed based on plasma factor VIII levels, patient and family referral for genetic screening and counseling for factor VIII gene mutation analysis to establish carrier status. The outlook for most hemophilia A is guarded. Repeated transfusions of blood products and related factors is not being event. Additionally, these patients are prone to bleeding, which can be life-threatening. The pharmacist should perform a thorough medication check, looking for any agents that might precipitate bleeding, as well as verifying dosing of clotting factors and any other medications (e.g., pain control). Nursing can provide patient counseling, reported any concerns to the managing clinician, and monitoring patient progress and status. These interprofessional examples demonstrate how this approach can optimize patient outcomes.
Suggested Reading
- Overview of Coagulation System
http://www.womenshealthsection.com/content/obsnc/obsnc015.php3 - Vitamin K Deficiency Bleeding
http://www.womenshealthsection.com/content/obsnc/obsnc014.php3 - Managing von Willebrand Disease (VWD) in Women
http://www.womenshealthsection.com/content/gyn/gyn036.php3
References
- Schramm W. The history of hemophilia – a short review. Thromb Res 2014;134(Suppl 1):S4-9
- Zimmerman B, Valentino LA. Hemophilia: in review. Pediatr Rev 2013;34(7):289-294
- Bertamino M, Riccardi F, Banov L, et al. Hemophilia care in the pediatric age. J Clin Med 2017;19(6):54 available @ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5447945/ Last accessed 2 January 2023
- Berntorp E, Shapiro AD. Modern haemophilia care. Lancet 2012;379:1447-1456
- Tonbary YA, Elashry R, Zaki MS. Descriptive epidemiology of hemophilia and other coagulation disorders in manouri, Egypt: retrospective analysis. Mediterr J Hematol Infect Dis 2010;2(3):e2010025
- Peyvandi F, Garagiola I, Young G. The past and future of hemophilia: diagnosis, treatments, and its complications. Lancet 2016;388:187-197
- Pipe SW, Montgomery RR, Pratt KP, Lenting PJ, Lillicrap D. Life in the shadow of a dominant partner: the FVIII-VWF association and its clinical implications for hemophilia A.
- Michaux G, Cutler DF. How to roll an endothelial cigar: the biogenesis of Weibel=Palade bodies. Traffic 2004;5(2):69-78
- Peyvandi F, Garagiola I, Young G. The past and future of hemophilia: diagnosis, treatments, and its complications. Lancet 2016;388(10040):187-197
- Zimmerman B, Valentino LA. Hemophilia: in review. Pediatr Rev 2013; 34(7):289-294
- Bertamino M, Riccardi F, Banov L, et al. Hemophilia care in pediatric age. J Clin Med 2017;19
- Schep SJ, Boes M, Schutgens REG, van Vulpen LFD. An update on the ‘danger theory’ in inhibitor development in hemophilia A. Expert Rev Hematol 2019;12(5):335-344
- Salen P, Babiker HM. Hemophilia A. Treasure Island (FL): StatPearls Publishing 2021 Jan. 2021 Jul 21
- Preijers T, Schutte LM, Kruip MJHA, et al. Strategies for individualized dosing of clotting factor concentrates and desmopressin in hemophilia A and B. Ther Drug Monit 2019; 41(2):192-212
- Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med 2017;377:809-818
- Schramm W. The history of haemophilia – a short review. Thromb Res 2014;134(Suppl 1):S4-9
- Iorio A, Stonebraker JS, Chambost H, et al. Data and Demographic Committee of the World Federation of Hemophilia. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Intern Med 2019;171(8):540-546
- Lowe GD, Ludlam CA. Less severe bleeding in hemophilia B than in hemophilia A. J Thromb Haemost 2008;6():1982-1983
- Stonebraker JS, Bolton-Maggs PH, Soucie JM, et al. A study of variations in the reported hemophilia A prevalence around the world. Haemophilia 2010;16(1):20-32
- Stobart K, Iorio A, Wu JK. Clotting factor concentrates given to prevent bleeding and bleeding-related complications in people with hemophilia A or B. Cochrane Database Syst Rev 2006;19(2): CD003429
- Djambas KC. Once-weekly prophylactic dosing of recombinant factor IX improves adherence in hemophilia B. J Blood Med 2016;7:275-282
- Nathwani AC, Davidoff AM, Tuddenham EGD. Gene therapy for hemophilia. Hematol Oncol Clin North Am 2017;31(5):853-868
- Chitlur M, Warrier I, Rajpurkar M, Lusher JM. Inhibitors in factor IX deficiency a report of the ISTH-SSC international FIX inhibitor registry (1997-2006). Haemophilia 2009;15:1027-1031
- Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost 2009;7(suppl 1):84-87
- Gidley GH, Holle LA, Burthem J, et al. Abnormal plasma clot formation and fibrinolysis reveal bleeding tendency in patients with partial factor XI deficiency. Blood Adv 2018;2(10):1076-1088
- Gerber GF, Klute KA, Chapin J, Bussel J. et al. Peri- and postpartum management of patients with factor XI deficiency. Clin Appl Thromb Hemost 2019;25:1076029619880262
- Salomon O, Steinberg DM, Seligshon U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia 2006;12(5):490-493
- Janbain M, Leissinger CA, Kruse-Jarres R. Acquired hemophilia A: emerging treatment options. J Blood Med 2015;6:143-150
- Holme PA, Brosstad F, Tjonnfjord GE. Acquired haemophilia: management of bleeds and immune therapy to eradicate autoantibodies. Haemophilia 2005;11(5):510-515
- Janbain M, Leissinger CA, Kruse-Jarres R. Acquired hemophilia A: emerging treatment options. J Blood Med 2015;6:143-150
- Windyga J, Baran B, Odnoczko E, Buczma A, et al. Treatment guidelines for acquired hemophilia A. Ginekol Pol 2019;90(6):353-364
- Duncan E, Collecutt M, Street A. Nijmegen-Bethesda assay to measure factor VIII inhibitors. Methods Mol Biol 2013;992:321-333
- 3Collins PW. Management of acquired hemophilia A. J Thromb Haemost 2011;9(suppl 1):226-235
- Huth-Kuhne A, Baudo F, Collins P, Ingerslev J, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica 2009;94(4):566-575
- Zeng Y, Zhou R, Duan X, Long D. Rituximab for eradicating inhibitors in people with acquired haemophilia A. Cochrane Database Syst Rev 2016;7:CD011907
- Melchiorre D, Linari S, Manetti M, Romano E, et al. Clinical, instrumental, serological and histological findings suggest that hemophilia B may be less severe than hemophilia A. Hematologica 2016;101:219-225
- Witkop M, Guelcher C, Forsyth A, Hawk S, et al. Treatment outcomes, quality of life, and impact of hemophilia on young adults (aged 18-30 years) with hemophilia. Am J Hematol 2015;90 (Suppl 2):S3-S10
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS et al. Guidelines for the management of hemophilia. Haemophilia 2013;19:e1-e47
- Mancuso ME, Berardinelli L. Arteriovenous fistula as stable venous access in children with severe haemophilia. Haemophilia 2010;16(Suppl 1):25-28
- Cuesta-Barriuso R, Torres-Ortuno A, Perez-Alenda S, Jose Carrasco J, et al. Sporting activities and quality of life in children with hemophilia: an observational study. Pediatr Phys Ther 2016;28(4):453-459
- Croteau SE. Hemophilia A/B. Hematol Oncol Clin North Am 2022;36(4):797-812
Опубликован: 14 January 2023
Dedicated to Women's and Children's Well-being and Health Care Worldwide
www.womenshealthsection.com