Программа скрининга новорожденных в СШАБюллетень WHEC Практика и клинической управления для медицинских работников. Образование гранта, предоставленного здоровья женщин и образовательный центр (WHEC). Newborn screening is the largest screening program in the United States with approximately four million newborns screened yearly. It is a mandated public health program designed for the identification of disorders in children. It is designed to provide rapid diagnosis and allow early therapy for specific metabolic, infections, and other genetic disorders for which early intervention reduces disabilities and death. This important practice typically occurs before the development of signs or symptoms of disease. Newborn screening programs are comprised of a complex, integrated clinical service of education, screening, diagnosis, follow-up, evaluation, and often long-term management. The goal of these essential public health programs is to decrease morbidity and mortality by screening for disorders for which early intervention will improve neonatal and long-term health outcomes for the individual. Most of the disorders screened through these programs have no clinical findings at birth. In 2006, the Centers for Disease Control and Prevention (CDC) estimated that approximately 68% of children identified as screen-positive by newborn screening in the United States were affected by galactosemia, the major hemoglobinopathies, phenylketonuria (PKU), and congenital hypothyroidism (1). The remaining one third were affected by one of the other disorders identified as recommended targets for newborn screening. Given that newborn screening typically occurs while the mother is immediately postpartum, obstetricians and health care providers caring for women during this critical time are uniquely positioned to give basic information to women before delivery and to answer questions during the antenatal and postpartum period. The purpose of this document is make resources regarding newborn screening available to the healthcare providers and the patients. Newborn screening programs in the United States are developed and managed at the state level, and operate through collaborations between public health programs, laboratories, hospitals, pediatricians, subspecialists, and specialty diagnostic centers. Their functions include the initial screening of all newborns, identifying screen-positive neonates, diagnosing conditions, communicating with families, ensuring that affected children are referred to treatment centers, following long-term outcomes, and educating physicians and the public according to individual state guidelines. Although current state requirements vary, the results of surveys and focus groups of expectant parents demonstrate that, women and their families would like to receive information about newborn screening, during their prenatal care. Newborn Blood Spot ScreeningAlmost all states in the United States have adopted the 2010 Recommended Uniform Screening Panel suggested by the U.S. Secretary of Health and Human Services' Advisory Committee on Heritable Disorders in Newborns and Children. The list of recommended conditions for newborn screening is continually being evaluated (2). The National Newborn screening and Global Resource Center maintains a current list of conditions screened for each state in the United States (3). Newborn blood spot screening programs are developed and managed at the state level, and operate through collaborations between public health programs, laboratories, hospitals, pediatricians, subspecialists, and specialty diagnostic centers. A comprehensive screening program includes the following components:
Every birthing facility establish routines to ensure that all newborn infants are screened in accordance with state law. States test newborn infants primarily through blood samples collected from heel pricks that are placed on a special filter paper. Umbilical cord blood is never an appropriate specimen because it will be inaccurate for detection of disorders in which metabolite accumulation occurs after birth and after the initiation of feeding. Newborn screening blood specimens are ideally collected between 24 hours and 48 hours of age and sent to the designated state newborn screening laboratory as soon as possible. If the initial specimen is obtained before the infant is 24 hours old, most states recommend that a second specimen be obtained to decrease the probability of false-negative test results for disorders with metabolite accumulation (e.g. phenylketonuria) or false-positive test results (e.g. hypothyroidism) because of early testing. Some states mandate, or strongly recommend, that an additional newborn screening blood specimen be collected on all infants at 10 - 14 days of age, to reduce the chance of missed identification of infants with clinically significant disorders. Regardless of the screening test results, diagnostic testing should be performed if clinically indicated, as some affected infants will not be identified because of individual or biologic variations, very early discharge, or administrative or laboratory error. An adequate dried blood specimen must be provided to the laboratory for accurate testing. Limitations for obtaining an adequate specimen include infants who receive a transfusion or total parenteral nutrition, are sick, or are preterm. The Clinical and Laboratory Standards Institute recommends that screening of preterm and sick newborn infants be performed on admission to neonatal intensive care unit (NICU), at 48 - 72 hours of age, and again either at 28 days of age or discharge (whichever is sooner). For these infants, nurseries should develop protocols that comply with state regulations. The responsibility for transmitting the screening test results to the health care providers should rest with authority or agency that performed the test. However, primary care providers must develop policies and procedures to ensure that newborn screening is conducted, that results are transmitted to them in a timely fashion, and that the information is documented in the medical record. Primary care providers also must develop strategies to employ should these systems fail. HistoryNewborn genetic screening was initiated more than 50 years ago by Dr. Robert Guthrie of New York State when he developed a simple blood test to detect elevated levels of phenylalanine in newborns with phenylketonuria (4). Phenylketonuria (PKU) is an autosomal-recessive metabolic condition in which individuals lack the hepatic enzyme phenylalanine hydroxylase and develop elevated levels of phenylalanine causing seizures and significant developmental delay. The blood test used for detection of PKU, a bacteria inhibition assay, relied on the finding that high phenylalanine levels prevented bacteria from growing. If a diagnosis was made through this test, mental retardation could be avoided by placing affected newborns on a special diet lacking phenylalanine. Newborn screening for PKU was subsequently instituted in all 50 states and the District of Columbia in the United States in 1960s. The success of PKU screening established the basis for all forms of newborn screening. Specifically, it met the original criteria for screening for inborn errors of metabolism set forth by Wilson and Jungner in 1968 (5). States have added additional conditions to their newborn screening panels based on these criteria, including congenital hypothyroidism, congenital adrenal hyperplasia, homocystinuria, galactosemia, maple syrup urine disease, biotinidase deficiency, and hemoglobinopathies such as sickle cell anemia. Principles of Early-Disease Detection (6):
The list of recommended conditions for newborn screening programs is continually being evaluated. The introduction into newborn screening programs of tandem mass spectrometry, a method of measuring the molecular mass of a sample, has made it possible to detect many more disorders. Additional disorders identified using tandem mass spectrometry are included in some states' newborn screening panels. These secondary targets are believed to be clinically significant, but they have an unclear natural history or lack an appropriate medical therapy that affect long-term outcome (7). Informed ConsentAll U.S. states, U.S. territories, and the District of Columbia have individual newborn screening programs. Statutes and regulations are variable across states. Some programs require that parents be given the option to provide consent. Other states require the parent (or guardian) to provide consent if identifiable information will be disclosed outside the program. Penalties exist for violating newborn screening regulations in several states (8). Recommended Uniform Newborn Screening Panel of Core ConditionsThis summary is based on recommendations form a multidisciplinary team of experts, included in uniform panel of 29 core conditions for which screening is currently recommended for all newborns (9). As of November 2016: 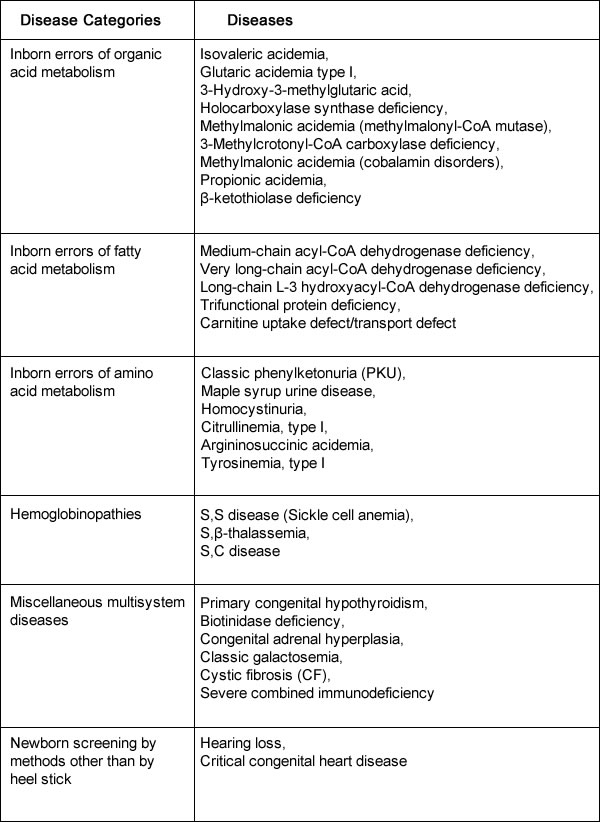 Although the original focus of newborn screening was on conditions that affect the central nervous system, the core panel now tests for five main categories of disorders:
Tandem mass spectrometry is used to test for disorders of amino acid, fatty acids, and organic acids. High-performance liquid chromatography is often used to test for several different types of hemoglobinopathies, and a variety of other techniques are used to test for other main conditions. Amino Acid DisordersAmino acid disorders are inherited metabolic disorders, which, because of an interruption of amino acid metabolism, cause a build-up of toxins. General symptoms of amino acid disorders include poor feeding, lethargy, hypotonia, seizures, mental retardation and developmental regression, unusual odors, and growth failure. An example of such a disorder would be phenylketonuria (PKU). The incidence of these disorders ranges from 1:25,000 (phenylketonuria) to 1:100,000 (maple syrup urine disease, citrullinemia, tyrosinemia type I, among others) (10). Most of these disorders are autosomal-recessive and are therefore unlikely to be identified by a family history. Newborns in the United States are screened for PKU, a metabolic disorder that when left untreated is characterized by elevated blood phenylalanine (phe) levels and severe mental retardation. An estimated 3,000 to 4,000 U.S. born women of reproductive age with PKU have not gotten severe mental retardation, because as newborns their diets were severely restricted in the intake of protein-containing foods and were supplemented with medical foods (e.g. amino-acid-modified formula and modified low-protein foods (11). When women with PKU do not adhere to their diet before and during pregnancy, infants born to them have a 93% risk for mental retardation and a 72% risk of microcephaly (11). These risks result from the toxic effects of high maternal blood phe levels during pregnancy, not because the infant has PKU. The restricted diet, which should be maintained for life, often is discontinued during adolescence. Maintaining the recommended dietary control of blood phe levels before and during pregnancy are crucial. Treatment generally consists of medications as well as a low-protein diet. Organic Acid DisordersOrganic acid disorders are each associated with a specific enzyme deficiency, which leads to an accumulation of blood vessels of the specific organic acid. These disorders have a variable age of onset depending on the condition. Increased levels of organic acids can cause lethargy, failure to thrive, vomiting, seizures, developmental delay, and coma. Most require specific protein restrictions and nutritional supplements. Propionic acidemia would be an example of such a disorder. Most are autosomal-recessive in inheritance and are therefore unlikely to be identified in a family prenatally. The incidence of these ranges from 1:75,000 to 1:100,000 Fatty Acid DisordersFatty acid disorders are inherited metabolic conditions that decrease energy metabolism because of an accumulation of fatty acid metabolites. Affected individuals have an impaired ability to metabolize fats. Specific enzymes such as medium chain acyl co-A dehydrogenase deficiency affect the fatty acid metabolic pathway. The classic clinical presentation of children with these disorders is one of an apparently health child who, when undergoing periods of prolonged fasting or increased energy demands, develops unexplained lethargy, vomiting, seizures and becomes nonresponsive. Affected children require regular feeding to avoid periods of relative starvation because they have an impaired ability to metabolize fats. Most fatty acid disorders have an autosomal-recessive inheritance pattern. Therefore, it is unlikely that a family history (unless consanguineous) would identify these disorders within a family. The incidence ranges from 1:25,000 to 1:100,000 HemoglobinopathiesHemoglobinopathies are relatively common conditions with variable severity, ranging from mild anemias to damage to organ systems, infections, and significant pain. They may be the result of structural abnormalities in the hemoglobin molecule in disorders such as sickle cell anemia. The clinical manifestations may also be caused by an inadequate production of hemoglobin caused by a-thalassemia. Although the thalassemias are classically found to be most common in those of Mediterranean, Asian, African, or Indian descent, given the increasing presentation of admixed populations from various racial and ethnic groups. These disorders can be identified in all ethnic groups. Therapies may include support for persistent anemias including blood transfusions, pain management, prophylactic antibiotics, vaccinations such as Pneumovax for those with sickle cell anemia, and medical screening to assess end-organ damage resulting from these disorders. Most have autosomal-recessive inheritance. The incidence ranges vary greatly depending on the disorder and ethnicity. For example, the risk for sickle cell disease ranges from 1:400 for those of African American descent to 1:5,000 for those of other ethnic background. Miscellaneous Disorders in Newborn ScreeningCystic Fibrosis (CF) After sickle cell anemia, cystic fibrosis (CF) is the second most common inherited life-shortening disease of childhood onset in the United States. Treatment depends on the severity of presentation, which is highly variable. The severe manifestations of CF include pulmonary disease, failure to thrive, and pancreatic insufficiency. Milder forms can present as male infertility with congenital bilateral absence of vas deferens or nasal polyps. Patients with CF have mutations in the CF transmembrane conductance regulator gene found on chromosome 7. Although the incidence of CF differs by ethnicity (given that different mutations segregate with varying ethnic groups), the overall birth prevalence of CF in the United States is approximately 1:3,500 births (12). All women who present for preconception or prenatal care should be offered screening for CF. Current standards suggest that a panel of at least 23 mutations be used for CF screening during pregnancy (13). Newborn screening evaluates functional defects in the CF protein by initiating screening with immunoreactive trypsinogen concentration. Immunoreactive trypsinogen levels are elevated in children with CF, presumably from the leaking of this protein into the circulation after exocrine pancreatic injury. If an initial immunoreactive trypsinogen level is elevated, some states repeat the dried blood spot test in 2-3 weeks from the original sample; most others perform mutation analysis from the blood spot for a set of CF mutations common to the ethnic groups in their individual state. In programs that perform mutation analysis, two mutations identified on the blood spot confirm the diagnosis of CF. If only one mutation is identified, the child is sent for a sweat test as the definitive assessment of CF, because the second mutation may not be identified by the state's panel (14). From states that perform two immunoreactive trypsinogen tests, sweat testing is also the definitive confirmatory test. Sweat testing can be reliably performed after 1 week of life. Currently, a sweat chloride level of 60 mmol/L is diagnostic of CF; a value ranging from 30 mmol/L to 59 mmol/L is a borderline result that requires a repeat test, DNA analysis (if not previously performed), or both (4),(14). Approximately 5% of individuals will have persistent borderline sweat tests and it is unclear what the long-term outcome is for these individuals. Newborns with CF who have pancreatic insufficiency in the first few weeks of life are at risk of severe nutritional complications. Pancreatic enzyme replacement therapy, fat-soluble vitamin supplements, and salt replacement are initiated immediately after the diagnosis is made in pancreatic-insufficient patients. Additional therapy relates to symptoms and presentation but may include nutritional supplementation, pulmonary therapy, antibiotics, and enzyme replacement for pancreatic disorders. CF is inherited in an autosomal-recessive manner. Biotinidase Deficiency Biotinidase deficiency prevents the recycling of the vitamin biotin. It can result in seizures, infections, hearing loss, and mental retardation; if untreated. It can result in coma and death. Treatment with daily biotin supplementation completely prevents these symptoms. The inheritance pattern is autosomal-recessive with an incidence of greater than 1:75,000 births (15). Congenital Adrenal Hyperplasia Congenital adrenal hyperplasia can be caused by various enzyme deficiencies. The most common form is the result of an enzyme deficiency of 21 hydroxylase, which results in impaired adrenal synthesis of cortisol from cholesterol. The salt-wasting form of 21 hydroxylase deficiency has an incidence of 1:15,000 live births. This form causes fetal virilization in affected female fetuses. Virilization of the genetically female fetus often results in ambiguous genitalia; excess androgens do not produce anatomic changes in male offspring. Because newborns with salt-wasting form can have life-threatening crises, rapid identification of the 21-hydroxylase form through newborn screening is essential. Management includes glucocorticoid replacement as well as management of the virilized female. All forms are inherited as autosomal-recessive conditions; the incidence is greater than 1:25,000 births for all forms of this enzyme disorder. Congenital Hypothyroidism Congenital hypothyroidism may result from and absent thyroid gland or from failure of the thyroid to either develop or function properly. It can cause severe growth delay and mental retardation because of inadequate of absent thyroid hormone in the newborn. Therapy requires life-long thyroid replacement, which can be taken orally. If treatment begins within the first month of life, development is usually normal. Although most cases are sporadic, approximately 20% of these are inherited, mostly in an autosomal-recessive manner; some autosomal-dominant cases have been described. The incidence is estimated at greater than 1:5,000 births (16). Galactosemia Galactosemia is caused by a deficiency in the galactose-1 phosphate uridyltransferase enzyme, which results in impaired galactose metabolism. This liver enzyme is needed to convert galactose into glucose for energy metabolism. The accumulation of galactose causes the clinical presentation of failure to thrive, infection, cataracts, liver failure, mental retardation, and death. Dietary intervention is designed to restrict galactose and has variable outcomes. A milder presentation of galactosemia is known as the "Duarte variant," which is often but not always detected by newborn screening. These children may have no sequelae or have milder findings than the severe form of galactosemia. Treatment of this form is controversial. Both types have an autosomal-recessive inheritance pattern. The incidence is greater than 1:30,000 births for classic galactosemia and approximately 1:16,000 for the Duarte variant (17). Hearing If undetected at birth, hearing impairment can affect speech and language acquisition, emotional and social development, and academic achievement. Without newborn hearing screening, most children are not identified as hearing-impaired until 2-3 years of age. The number of newborns born with significant permanent hearing loss is estimated at 1 to 3 per 1,000 with an estimated 3 per 1,000 with moderate hearing loss (18). Newborn hearing screening is typically performed by evoked otoacoustic emissions or by the auditory brain stem response measures. Otoacoustic emissions are designed to measure the cochlea's response to sound. To perform the test, a small probe is placed in the newborn's ear, sounds are introduced, and the response is recorded. If no response is noted, the newborn may have a hearing deficiency. To obtain auditory brain stem response measures, 3 surface electrodes are placed on the forehead, nape, and mastoid to detect waveforms recorded from stimuli given at 35 dB. The waveforms generated are compared with a standard. Delayed or absent waves are suggestive of a neurologic or cochlear defect. Newborn hearing screening defines permanent unilateral or bilateral hearing loss as 30 - 40 dB hearing level or greater across the frequencies of 500 - 40,000 Hz, which is the range that is essential for speech recognition and comprehension. Newborns who fail either screening test are referred for additional audiologic evaluation. Severe Combined Immunodeficiency (SCID) Babies born with Severe Combined Immunodeficiency (SCID) appear normal at birth but cannot fight infection. They may die before 1 year of age without medical treatment (19). If SCID is diagnosed early in life, before the onset of infection, a bone marrow transplant can successfully treat disorder. SCID is a primary immune deficiency syndrome with a severe defect in both the T and B lymphocytes. It causes an increased susceptibility to a variety of infections as well as failure to thrive. Children with untreated SCID have a life expectancy of approximately 2 years. It is the first newborn screening disorder that is DNA-based for identification. Currently in USA, approximately eight states screen for SCID; seven states have testing required by their state but have not yet implemented screening (20). Although it has been recommended as an addition to the core panel of screened disorders, it may be several years before it is adopted nationally. There are several forms of this disorder; it is most commonly inherited as an autosomal-recessive condition but also occurs with X-linked inheritance. The incidence is greater than 1:100,000 live births (20). Treatment consists mainly of early bone marrow transplant. Critical Congenital Heart Disease Congenital heart disease affects approximately 8:1,000 newborns and accounts for 24% of all infant deaths resulting from congenital birth defects (21). The subgroup of critical congenital heart disease is defined as heart disease with a severe, life-threatening presentation within the first year of life and is composed of seven conditions:
In September 2010, the Secretary's Advisory Committee on Heritable Disorders in Newborns and Children voted to add critical congenital heart disease to the core panel, recommending the use of pulse oximetry to identify critical congenital heart disease in newborns. The non-invasive pulse oximetry evaluation measures the percent of hemoglobin oxygen saturation and is a technique that is readily available and could be implemented in all newborn nurseries. Although this recommendation has been made, the committee also suggested that evaluation of protocols for implementation of the screening test and demonstration of improved health outcomes will need to be performed and that Health Resources and Services Administration should guide screening standards and infrastructure for this program. As of February 2012, only New Jersey was screening for critical congenital heart disease. Ethical, Legal, and Social IssuesThe medical screening and diagnostic testing of minors raises the consideration of substantial ethical, legal, and social concerns. Among these are issues regarding the benefits and risks of screening, the identification and management of both primary and secondary diseases (or targets), the psychological effect of false-positive screening for adult disease with more advanced genetic testing, consent requirements, the use of residual blood spots for research, and educational requirements as newborn screening expands and adopts new technologies (22). The original criteria for adding a condition to newborn screening panels suggested that an immediate, life-saving intervention needed to be available to warrant screening. This assumption has been challenged, and some groups have suggested a broader consideration of benefits (23). Secondary targets, which are conditions that can be easily identified by tandem mass spectrometry but for which no known therapies exist, generate complex issues. In addition, identifying secondary targets for which there are currently no known treatments can allow families to enroll their newborns in studies that can define the natural history of these conditions and potentially lead to therapies (24). As next-generation of sequencing becomes more efficient and less expensive, every newborn's entire genome sequence potentially could be available within 1 week of birth. How this information should be identified and reported will need to be determined (25). Newborn screening will therefore can screen for adult-onset conditions. For example, although there is no immediate need for a family to know that their newborn is a carrier of a BRCA1 or BRCA2 mutation associated with familial breast and ovarian cancer syndrome, testing a newborn is technically possible. Currently, these types of targets have not been considered for newborn screening; however, genetic predisposition to childhood-onset conditions such as multiple endocrine neoplasia type 2 may challenge this position when interventions during childhood such as prophylactic thyroidectomy have been shown to be lifesaving. The success of newborn screening has led to unanticipated consequences for obstetricians as the first generation of individuals who benefited from newborn screening enter their reproductive years. Newborns who were diagnosed with PKU and adhered to phenylalanine-free diets have led healthy lives and many relax their dietary restriction in late adolescence and adulthood. However, once the woman with PKU is planning a pregnancy or becomes pregnant, it is essential that she continue her strict phenylalanine-free diet so that her developing fetus does not suffer the neurologic consequences of elevated maternal phenylalanine levels. Identifying women with PKU and providing them with guidance during preconception care as well as offering carrier screening to their partners to determine their risk for an affected child are important roles for specialty obstetric and genetic services (26). Current and Future Challenges of Newborn ScreeningPublic health departments must continue to maintain newborn screening programs, to integrate all aspects of care from screening to medical therapy, and to introduce new screening tests. Advances in genomic medicine have the capacity to shift the focus of newborn screening away from the diagnoses that need immediate intervention and toward the screening of newborns for disorders that affect long-term health such as the propensity to develop coronary artery disease or some forms of cancer. This will cause significant organizational and financial stresses and may require a reassessment of the goals of newborn screening and complex evaluation and planning before implementation. The management of false-positive testing continues to be challenging for screening programs. Given the stress placed on families with false-positive screens, minimizing false-positive tests is important as new tests are being considered for inclusion. Furthermore, some children are not screened, either because of issues of illness or home birth or because of parents' refusal to screen their newborn for religious considerations, which is allowed in many states. Finally, although this is a broad public health program, there is limited public awareness about the value of newborn screening (27). Healthcare providers have an essential role in the success of newborn screening by informing pregnant women about this important public health program before delivery. SummaryIntegrating education about newborn screening into prenatal care allows parents to be prepared for having their child undergo screening as well as for receiving newborn screening test results. Furthermore, parents often view their care from prenatal management through pediatrics as a continuum of care without health care provider distinctions. This can be accomplished at different moments in prenatal care: 1) during the first-trimester new obstetric visit and include written or web-site information along with other patient education materials, 2) later in pregnancy with other educational information is routinely distributed, such as at the time of glucola or group B streptococcal screening in the third trimester, 3) during a discussion of past adverse pregnancy outcomes related to a positive newborn screening test result or birth defect, at the same time that options for prenatal or preimplantation genetic screening or diagnostic testing are considered. Newborn screening programs are state-based public health programs that provide all newborns in the United States with pre-symptomatic testing and necessary follow-up health care for a variety of medical conditions for which early intervention will improve neonatal and long-term health outcomes. All U.S. states and the District of Columbia have individual newborn screening programs with varying policies, statutes, and regulations. Newborn screening consists of blood-spot screening for metabolic and genetic conditions, hearing screening, and pulse oximetry screening for critical congenital heart defects. State health department's newborn screening programs vary as to the long-term disposition of samples for storage and research. RecommendationThe Women's Health and Education Center (WHEC) recommends that obstetric care providers make resources regarding newborn screening available to patients during pregnancy. Information can be disseminated through informational brochures, electronic sources, or through discussion during prenatal visits. Suggested Reading
References
|