Серповидно Сотовый и беременностьWHEC Практика бюллетень и клинической Управление медицинских работников.Образовательный грант предоставляется по охране здоровья женщин и образовательный центр (WHEC). Sickle cell disease refers to a group of autosomal recessive disorders involving abnormal hemoglobin (hemoglobin S). It is a devastating abnormality of red blood cells that results in circulatory impairment, tissue damage, infarctions, severe anemia, and life-threatening infections. Sickle cell disease affects between 70,000 to 100,000 Americans, mostly of African-descent, with a minority of Hispanic, southern European, Middle Eastern, and Asian Indian descent. Today, sickle cell disease is most often discovered during routine newborn screening. Although sickle cell disease is associated with major morbidity, more than 90% of children with sickle cell disease in the United States survive into adulthood. Compared to the general population, however, their life-spans are 2 or 3 decades shorter and limited by both acute and chronic morbidity. Acute complications of sickle cell disease include ischemic, vaso-occlusive (pain) crises, acute chest syndrome (which most closely resembles pneumonia, but may also result from fat embolism from bone marrow, intrapulmonary aggregates of sickle cells, atelectasis, or pulmonary edema), stroke, splenic sequestration, acute renal failure, and cholecystitis. Chronic complications include chronic pain, cholelithiasis, renal dysfunction, hypertension, pulmonary hypertension, and retinal problems (1). By the time they reach childbearing age, young women with sickle cell disease may have suffered many severe complications. Special circumstances during pregnancy appreciably increase morbidity among these women. The purpose of this document is to review sickle cell disease in pregnancy and to provide recommendations, screening and clinical management during prenatal and puerperium. Genetic screening can identify couples at risk for offspring with sickle cell disease and other hemoglobinopathies and allow them to make informed decisions regarding reproduction and prenatal diagnosis (2). Individuals with African, Southeast Asian, and Mediterranean ancestry are at a higher risk for being carriers of hemoglobinopathies and should be offered carrier screening. Ethnic groups considered to be at low-risk for hemoglobinopathies include northern Europeans, Japanese, Native Americans, Inuit (Eskimo), and Koreans. If both parents are determined to be carriers, genetic counseling is recommended. It should be noted ethnicity is not always a good predictor of risk because individuals from at-risk groups may marry outside their ethnic group. BackgroundSickle cell disease was first linked to an abnormality of red blood cells by Dr. Ernest Irons and Dr. James Herrick in 1971. By the 1920s, enough experience had accumulated about the disease to call it sickle cell anemia, due to abnormally shaped red blood cells that resemble the classic “sickle” or crescent shaped found on peripheral blood smears. By the late 1940s, sufficient biochemical and genetic data has accumulated for Linus Pauling to call it the first molecular disease (3). Genotypes include homozygous hemoglobin SS (Hb SS) and compound heterozygous conditions, such as hemoglobin SC (Hb SC), hemoglobin Sβ°-thalassemia (Hb Sβ°-thal), hemoglobin Sβ+-thalassemia (Hb Sβ+-thal), and several rare genotypes. The most prevalent phenotype is Hb SS, which accounts for about 70% of cases and is identified as sickle cell anemia. The Hb SC variant accounts for most of the remaining cases. Hb Sβ°-thal, due to its similar phenotype, is also identified as sickle cell anemia. These two genotypes are associated with the most severe manifestations of sickle cell disease. The gene defect in Hb S causes hemoglobin to form ling chains or polymers that deform red blood cells, damage their membranes, and alter the configuration of their cell surface molecules. These deformed red blood cells are stiff and angular and can be trapped with leukocytes to block circulation, causing inflammation, tissue damage, and infarctions in vital organs including the eyes, brain, heart, lungs, liver, spleen, kidneys, bones, and during pregnancy, the placenta. Early removal from the circulation and hemolysis lead to a very short life span of the sickled red blood cells (16 to 20 days, compared with 120 days in normal red blood cells). Hemolysis results in release of free hemoglobin that damages the endothelium, causing further red blood cell adhesiveness and blockage of small blood vessels (4). Sickle Cell DiseaseHemoglobin S (Hb S) differs from the normal hemoglobin A (Hb A) because of a single nucleotide substitution of thymine for adenine in the β-globin gene; this alteration causes a substitution of valine or glutamic acid in the number six position of the β-globin polypeptide. Asymptomatic individuals with heterozygous Hb S genotypes (carriers) are said to have sickle cell trait. The most severe form of the disease, Hb SS (homozygous Hb S), is called sickle cell anemia. Sickle cell disease occurs most commonly in people of African origin. Approximately 1 in 12 African Americans has sickle cell trait (5). 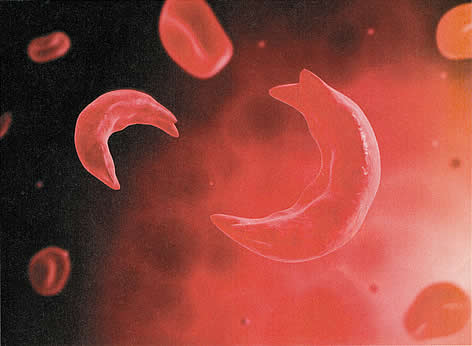 Figure 1: A microscopic view of sickled red blood cell One in every 300 African-American newborns has some form of sickle cell disease, and approximately 1 in 600 has sickle cell anemia. Hb S also is found in high frequency in other populations such as Greeks, Italians, (particularly Sicilians), Turks, Arabs, Southern Iranians, and Asian Indians (5). By the time they reach childbearing age, young women with sickle cell disease may have suffered many severe complications. Clinically apparent stroke occurs in 11% of those with sickle cell disease by age 20 and in 24% by age 45 (6). Although the condition is not usually regarded as a thrombophilia, 25% adults with sickle cell disease have experienced venous thromboembolism (VTE) by a median age of 30, which is comparable to the rate in adults with high-risk thrombophilias (6). Sickle Cell TraitAbout 8% of African Americans are heterozygous for the sickle cell gene. Sickle cell trait should not be considered a deterrent to pregnancy on the basis of increased risks to the mother. The risk of urinary tract infection is twice as high. Conflicting data exist regarding an association (increased or decreased) risk with pregnancy-induced hypertension. It appears that sickle cell trait does not unfavorably influence the frequency of abortion, perinatal mortality or low birth weight. The probability of a serious sickle cell hemoglobinopathies in offspring of women with sickle cell trait is 1 in 4 whenever the father carries a gene for abnormal hemoglobin or for β-thalassemia (7). Prenatal diagnosis of sickle cell disease through amniocentesis or chorionic villus sampling (CVS) is now available. CLINICAL FEATURESThe classical clinical feature of patients with sickle cell disease is seen under conditions of decreased oxygen tension, in which the red blood cells become distorted into various shapes, some of which resemble sickles. The distorted red cells lead to increased viscosity, hemolysis, and anemia and a further decrease in oxygenation. When sickling occurs within small blood vessels, it can cause “logjams” that can interrupt blood supply to vital organs (vaso-occlusive crisis). Repeated vaso-occlusive crises result in widespread microvascular obstruction with interruption of normal perfusion and function of several organs, including spleen, lungs, kidneys, heart, and brain. Adults with Hb SS are functionally asplenic, have undergone auto-splenectomy by adolescence. Absence of spleen contributes to the increased incidence and severity of infection in patients with sickle cell disease. The most significant threat to patients with sickle cell disease is acute chest syndrome. Chest syndrome is characterized by a pulmonary infiltrate with fever that leads to hypoxemia and acidosis. The infiltrates are not infectious in origin but rather are due to vaso-occlusion from sickling or embolization of marrow from long bones affected by sickling (5). The diagnosis of hemoglobinopathies, including sickle cell disorders, is made by hemoglobin electrophoresis. In the homozygous form, nearly all the hemoglobin is Hb S with small amounts of Hb A2 and Hb F. Heterozygous sickle cell trait (Hb AS) is identified by a larger percentage of Hb A and an asymptomatic course. Solubility tests (Sickledex) alone are inadequate for diagnosis of sickle cell disorders because they cannot distinguish between the heterozygous AS and homozygous SS genotypes. In addition, they fail to detect other pathologic variants such as Hb C trait, β-thalassemia trait, Hb E trait, Hb B trait, and Hb D trait. The only established disease-modifying therapies are chronic transfusion and hydroxyurea. The latter is strongly recommended for adults with 3 or more severe vaso-occlusive crises per year, pain or chronic anemia interfering with daily activities, or severe or recurrent episodes of acute chest syndrome. Hydroxyurea therapy is also suggested for adolescent without regard to symptoms. Long-term transfusion therapy is used to prevent stroke in children with abnormal transcranial Doppler velocity. Potential consequences of long-term transfusion therapy are alloimmunization and iron overload. Because transfused blood contains iron that circumvents the normal pathways of iron regulation, excess iron can accumulate in tissues and can become pathological. Chelation therapy can be used to remove excess iron in patients with evidence of iron overload (1),(8). PRECONCEPTION COUNSELINGCouples contemplating pregnancy should be aware of the increased risks to a woman’s health of sickle cell disease during pregnancy. Prior to pregnancy, the physician should assess a woman’s overall health and her potential risks during pregnancy. She and her partner should be counseled accordingly. Some studies have found poorer outcomes in women with sickle cell anemia as opposed to other sickle cell disease genotypes, but other studies have found no differences. It is the experience of the Center that a history of frequent hospitalizations and/or episodes of acute chest syndrome correlates with poorer overall health and increased risks during pregnancy. An assessment of urine protein and a retinal examination should be performed (9). Hypertension should be treated to lower systolic blood pressure to <140 mm Hg and diastolic blood pressure to <90 mm Hg. Some experts recommend that all sickle cell disease patients be screened for pulmonary hypertension with Doppler echocardiography (10). At a minimum, any woman with signs of pulmonary hypertension should undergo Doppler echocardiography. Women with significant pulmonary hypertension should be counseled that pregnancy is contraindicated and that if it occurs, termination should be considered (11). All patients with sickle cell disease should receive immunizations according to the Advisory Committee on Immunization Practices (ACIP), but with particular attention to the immunizations recommended for people who have functional asplenia. These include pneumococcal, haemophilus influenza type b, and meningococcal vaccinations. A ferritin level should be checked before prescribing any iron. A serum ferritin level >1,000 ng/mL is suggestive of iron overload. The American College of Obstetricians and Gynecologists (ACOG) recommends 4 mg of folate per day during pregnancy (7). Women with sickle cell disease who have been transfused should be tested for red blood cell alloantibodies. If a woman had red blood cell alloantibodies and the antibodies are known to cause hemolytic disease in the fetus or newborn, her partner should also be tested for the corresponding red blood cell antigen(s). If the partner tests positive, the couple should be counseled about the risks of hemolytic disease, how it is monitored, and how it is treated. Couples should also be counseled about the potential fetal and neonatal effects of maternal medications, including chronic opioids. Angiotensin-converting enzyme (ACE) inhibitors, which are used to treat microalbuminuria in adults with sickle cell disease, should be discontinued. Hydroxyurea, which has caused birth defects in experimental animals who were given very high doses, has not been associated with an increased risk of birth defects in infants with in-utero exposure (12). Women with sickle cell disease are at risk of having a child affected with sickle cell disease if their partners have sickle cell disease, Hb S, β-thalassemia trait, or are carriers of another abnormal hemoglobin such as Hb C. Women with sickle cell disease whose partners’ sickle cell or thalassemia status is unknown should be referred for hemoglobin electrophoresis. Women whose partners carry one of the traits listed above can avoid an affected pregnancy by undergoing preimplantation genetic diagnosis. Alternatively, after spontaneous conception, prenatal diagnosis of sickle cell disease is possible by CVS in the first trimester or by amniocentesis in the second trimester. Couples who are at risk of having a child affected with sickle cell disease should be referred for genetic counseling. ISSUES DURING PREGNANCYMaternalDuring pregnancy, sickle cell disease poses problems to both mother and fetus. Maternal problems can arise from chronic underlying organ dysfunction such as renal disease or pulmonary hypertension, from acute complications of sickle cell disease such as vaso-occlusive crises and acute chest syndrome, and from pregnancy-related complications. In normal, iron-replete women, red blood cell mass should increase by 400-450 mL during pregnancy to support the 40%-50% increase in blood volume. This increase in red blood cell mass is not achievable in women with sickle cell disease. During pregnancy 50%-70% of women with sickle cell disease require at least one hospitalization and 30%-40% require transfusion (13). In one cohort, women with sickle cell disease were hospitalized an average of 6 days during pregnancy (13). Women with sickle cell disease not only require transfusion, but also have frequently been transfused previously, and according to published reports, 20% to 50% of sickle cell disease patients all alloimmunized (14). Limited blood products are available to women who are alloimmunized and need transfusion, but the most serious consequence is the maternal risk of developing a delayed hemolytic transfusion reaction, which can be life-threatening. Another potentially life-threatening problem is pulmonary hypertension. If affects 6%-11% of sickle cell disease patients (10), and is especially morbid in pregnancy due to the increased cardiopulmonary demands of gestation. Although maternal mortality from pulmonary hypertension was previously reported to be 30%-50%, mortality remains in the range of 16% (11). Perhaps because of underlying renal disease, hypertension, or placental ischemia, women with sickle cell disease are more likely to experience preeclampsia and eclampsia (15). They are more likely to experience VTE, infections (urinary tract infections, pneumonia, sepsis, and postpartum infections), acute renal failure, and death (16). In the United States, the maternal mortality rate is approximately 10 times higher than it is for women without sickle cell disease (17). Similarly increased rates are seen in other countries. FetalFetal concerns for women with sickle cell disease include the consequences of utero-placental insufficiency, alloimmunization, and opioid exposure. Studies from multiple cohorts, including large population studies, have documented the increased risk of fetal growth restriction, preterm delivery, and stillbirth (18). Fetal growth appears to start normally, then lag after 25 weeks’ gestation (19). When fetal growth restriction occurs, it has been described as asymmetric. Women with sickle cell disease who have formed antibodies to fetal red blood cell antigens that are associated with hemolytic disease in the fetus or newborn and who are alloimmunized, are at risk of having an anemic fetus or a stillbirth (20). Although there are no data specifically for infants born to mothers with sickle cell disease, in a retrospective cohort study of infants born at Mayo Clinic between 1998 and 2009, neonatal withdrawal syndrome occurred in 5.6% of infants exposed to chronic narcotic use in-utero (21). MANAGEMENT DURING PREGNANCYDuring pregnancy, evaluation should include consultation with a hematologist and a maternal-fetal medicine specialist. Pertinent history includes the patient’s genotype, history of transfusion, hospitalizations, episodes of acute chest syndrome, vaso-occlusive crises, Doppler echocardiography, stroke, VTE, cholecystectomy, status of spleen, renal evaluations and immunizations. Issues not addressed before conception should be addressed early in pregnancy. As assessment of iron store is usually made with the initial complete blood count (CBC). Most patients with sickle cell disease have elevated serum ferritin levels and should not receive additional iron supplementation, but should continue to receive folic acid. Pregnant patients with sickle cell disease need increased prenatal folic acid supplementation. The standard 1 mg of folate in prenatal vitamins is not adequate for patients with hemoglobinopathies; 4 mg per day of folic acid should be prescribed because of the continual turnover of red blood cells. The most common cause of recurrent morbidity in Hb SS disease is painful crisis. If possible, precipitating factors, such as cold environment, heavy physical exertion, dehydration, and stress, should be avoided. Hydroxyurea has been shown to reduce the frequency of painful crises in non-pregnant patients with severe sickle cell disease. However, the use of hydroxyurea is not recommended during pregnancy because it is teratogenic. Hydroxyurea therapy, chelating agents, and ACE inhibitors should be discontinued, although only ACE inhibitors have been associated with congenital anomalies. Women with sickle cell disease are at high-risk of preeclampsia and both arterial thromboembolism and VTE. Therefore, it is reasonable to initiate low-dose aspirin after the first trimester (22). Women with a history of VTE should receive anticoagulation with low-molecular-weight heparin (LMWH) during pregnancy and the postpartum period. Care during pregnancy includes maternal and fetal surveillance. Monthly CBCs are typically performed to monitor for severe anemia. In the only randomized controlled trial published to date, prophylactic transfusion was associated with a decreased risk of pain crises and severe anemia, but no difference was observed in pregnancy outcomes (23). Other therapies that have shown promise in reducing vaso-occlusive crises and would potentially be safe in pregnancy (such as propranolol and LMWH) have not been confirmed in multicenter trials and have not been studied in pregnancy (24). Risks of adverse fetal outcomes are reduced, but not eliminated, with fetal surveillance, which may lead to planned early delivery. Fetal surveillance should include, at a minimum, serial ultrasonography for fetal growth every 3-4 weeks starting in the third trimester with initiation of antepartum fetal testing at 32 weeks’ gestation or sooner if indicated. A contributing factor to the increased incidence of stillbirth is utero-placental insufficiency, but sudden and unpredictable stillbirths do occur. Some experts recommend induction of labor after 37 weeks’ gestation (8). Plans for the mode of delivery should be according to obstetric indications while recognizing that the rate of cesarean delivery is higher in women with sickle cell disease (25). An anesthesiologist should be consulted in the third trimester or during any hospitalization. Although prophylactic transfusion was not shown to improve pregnancy outcomes in patient with sickle cell disease, preoperative transfusion therapy to increase hemoglobin levels to 10 g/dL is strongly recommended in patients with sickle cell anemia ( Hb SS and Hb Sβ°-thal), and should be considered in patients with other genotypes (1). When a transfusion is clinically indicated in the patient with sickle cell disease, the objective is to lower the percentage of Hb S to approximately 40% while simultaneously raising the total hemoglobin concentration to about 10 g/dL (7). Since the need for cesarean delivery with sickle cell disease are at high risk of cesarean delivery, it may be reasonable to aim for this total hemoglobin concentration in the last month of pregnancy. Controversy exists regarding the role of prophylactic blood transfusion in management of sickle cell disease in pregnancy. By limiting transfusion to situations in which it is clinically indicated, patients are not subjected to the increased risk for alloimmunization, viral infections, and iron overload. Major complications (e.g. worsening anemia; intrapartum complications such as hemorrhage, septicemia, and cesarean delivery; painful crisis; and chest syndrome) may require intervention with an exchange transfusion. There is no consensus regarding the exact hematocrit value below which transfusion should be considered. However, when a transfusion is clinically indicated in the patient with sickle cell disease, the objective is to lower the percentage of Hb S to approximately 40% while simultaneously raising the total hemoglobin concentration to about 10 g/dL. Hemoglobin levels and the percentage of Hb S should be monitored serially during the remainder of the pregnancy to determine the need for subsequent transfusions. Women with sickle cell disease have a risk of thrombosis comparable to that in patients with high-risk thrombophilia. Women with sickle cell disease should have pneumatic compression devices during antepartum hospitalizations, during labor, and at the time of cesarean delivery. For women who are not already receiving anticoagulation for a history of VTE, some consideration should be given to prophylactic or low-dose anticoagulation during pregnancy and for 6 weeks postpartum. Stem-cell Transplantation for Sickle Cell DiseaseThe only curative therapy for sickle cell disease is allogeneic hematopoietic stem-cell transplantation. Several hundred patients with sickle cell disease, almost exclusively children, have received transplants with bone marrow cells from HLA-identical siblings. Reported overall survival after transplantation is 92% to 94% and event-free survival is 82% to 86% (26). Besides complications from bone-marrow-suppressive chemotherapy and from transplantation-related mortality, there is the possibility of graft failure and chronic graft-versus-host disease. The chronic organ damage that was sustained prior to the transplantation also may be reversible. The risk of toxicities and complications from transplantation is so high in adults that the procedure is not usually considered for them. Despite rare anecdotal reports of successful pregnancies in women after stem-cell transplantation, the vast majority lose fertility as a consequence of the chemotherapy. The rare pregnant patient with sickle cell disease who has undergone stem-cell transplantation should be evaluated and managed as are other women with sickle cell disease. This patient may be at reduced risk of vaso-occlusive crises, but will likely have been selected for transplantation due to some chronic organ damage and may have suffered additional tissue damage from bone-marrow-suppressive chemotherapy. Regimens typically include busulfan, which should prompt an evaluation of pulmonary function (26). MANAGEMENT OF VASO-OCCLUSIVE CRISESThere are no randomized trials on the safety and efficacy of interventions (transfusion, oxygen therapy, fluid replacement analgesia, and/or steroids) for treating pain crises during pregnancy (27). A pregnant woman with sickle cell disease who presents with pain should be evaluated for other complications, and pain should be treated promptly and aggressively. Non-steroidal drugs are contraindicated, so opioids should be used. Many women with sickle cell disease have a high tolerance for opioids, but they still need adequate and appropriate medication to control pain during vaso-occlusive crises in pregnancy. Opioids should not be withheld due to concerns for addiction. Transfusion should be administered only if there are other indications. Oxygen should be given if saturation is less than 95% by pulse oximetry. Incentive spirometry should be initiated for patients who are hospitalized for a vaso-occlusive crisis. Hormonal contraception may provide women with sickle cell disease with decreased menstrual blood flow and improved hemoglobin levels. VTE is the primary risk associated with combined hormonal methods containing estrogen. Intrauterine devices (IUDs) and implants carry risks associated with the insertion procedure, and sterilization carries risks associated with the surgical procedures. Nonetheless, these risks are trivial compared to the risks associated with pregnancy. Copper IUDs (as opposed to progestin IUDs) may increase bleeding, which is a more significant problem for women with sickle cell disease. Systematic reviews of the literature have demonstrated the safety of progestin-only contraception for patients with sickle cell disease (28). The management of pregnant women with sickle cell disease includes close observation with careful evaluation of all symptoms, physical findings, and laboratory studies. One rather common danger is that the symptomatic women may categorically be considered to be suffering from sickle cell crisis. As a result, ectopic pregnancy, cholecystitis, or other serious obstetric or medical problems that cause pain, anemia, or both may be overlooked. Intense sequestration of sickled erythrocytes with infarction in various organs may develop acutely, especially late in pregnancy, during labor and delivery, and early in puerperium. Infarction usually is accompanied by severe pain, and because in many cases the bone marrow is involved, intense bone pain is common. Individuals of African, Southeast Asian, and Mediterranean descent are at increased risk for being carriers of hemoglobinopathies and should be offered carrier screening, and if both parents are determined to be carriers, genetic counseling. A complete blood count and hemoglobin electrophoresis are appropriate laboratory tests for screening for hemoglobinopathies. Solubility tests alone are inadequate for screening because they fail to identify important transmissible hemoglobin gene abnormalities affecting fetal outcomes. Couples at risk for having a child with sickle cell disease or thalassemia should be offered genetic counseling to review prenatal testing and reproduction options. Prenatal diagnosis of hemoglobinopathies is best accomplished by DNA analysis of cultured amniocytes or chorionic villi. |