Evaluación del riesgo de cáncer hereditario en GinecologíaWHEC Boletín de la práctica clínica y directrices de gestión de los proveedores de atención médica. Educación subvención concedida por la Salud de la Mujer y el Centro de Educación (WHEC). Evaluación del riesgo de cáncer hereditario debe ser parte de la obstetricia de rutina y la práctica ginecológica. En la última década se ha producido un aumento de la conciencia de la predisposición hereditaria a una amplia gama de enfermedades, incluyendo cáncer, enfermedades del corazón y diabetes. Estos son todos los complejos trastornos de múltiples genes, pero la identificación de genes específicos asociados con una predisposición a estas condiciones ha permitido a los médicos para evaluar con mayor precisión el riesgo y prescribir las intervenciones preventivas. A pesar de que puede ser desconocido para muchos profesionales, el proceso de estratificación de riesgo de cáncer puede ser eficiente y eficaz. Usando la evaluación basada en un protocolo de susceptibilidad al cáncer, los factores de riesgo personales y familiares, y las pruebas genéticas, es posible crear perfiles de riesgo y estrategias de gestión que demuestran la reducción demostrado en la morbilidad y mortalidad del cáncer. Décadas antes de la prueba genética entró en la práctica, los médicos tenían una carga excesiva de cáncer de mama y de ovario, a menudo con casos de inicio temprano y el cáncer de mama masculino. A mediados de los años 1990 el análisis de ligamiento en estos mama y de ovario familias con cáncer permitieron la identificación de los genes BRCA1 y BRCA2 , los genes que funcionan normalmente en la reparación del ADN. Consejeros genéticos del cáncer utilizan el análisis de pedigrí y los indicadores clínicos para evaluar la probabilidad de que un paciente lleva una mutación hereditaria, para determinar las estrategias de experimentación asignados entre un conjunto cada vez más complejo de las pruebas disponibles en el mercado, y para interpretar los resultados y orientar a las recomendaciones médicas. El descubrimiento científico, una amplia conciencia pública y la disponibilidad general de la prueba genética clínica del BRCA1 y BRCA2 genes responsables de la mayoría de mama familiar y el cáncer de ovario, han llevado a las mujeres a presentar a las oficinas del médico solicita pruebas genéticas y cuestionar las implicaciones clínicas para ellos y sus familias. El propósito de este documento para revisar las recomendaciones actuales para las pruebas genéticas para la susceptibilidad a los cánceres, incluyendo ovario, trompa de Falopio, de mama, de endometrio, y cánceres de colon, debido a mutaciones heredadas en los BRCA los genes o en los genes de reparación asociados con el cáncer de colon sin poliposis hereditario (HNPCC) Síndrome. Historia familiar sigue siendo la piedra angular de la identificación del paciente. Etnicidad y raza son habitualmente evaluados como parte de la evaluación. Las aplicaciones prácticas de las pruebas genéticas para la susceptibilidad al cáncer tienen la capacidad de reducir la carga de los cánceres hereditarios por salvar vidas, la disminución de la morbilidad médicos, y reducir el estrés psicológico. También se aborda Gestión de portadores de la mutación, incluidas las indicaciones para las cirugías de reducción de riesgos, detección de cáncer y de seguimiento,. El estudio también se centra en las prácticas clínicas y el uso práctico de la genética clínica del cáncer. FondoHay numerosos síndromes heredados y que confieren riesgo de cáncer de mama, de ovario, y cánceres relacionados con el 5% a 15% de todos los cánceres de mama y de ovario causada por uno de estos síndromes de cáncer, herencia dominante autosómica (1),(2). Los genes asociados con síndromes de cáncer hereditario son todos transmitidos de manera autosómica dominante mendeliana, las historias de la familia para la maternidad y paternidad contribuyen por igual al riesgo del paciente y deben ser evaluados para determinar la idoneidad de las pruebas. Cabe señalar, sin embargo, que, aunque las mutaciones en los genes se heredan de una manera autosómica dominante, que se expresan de una manera recesiva (2). Un individuo que hereda una mutación de línea germinal que inactiva una copia del gen, en general, tiene una segunda copia intacta funcional del gen. Es sólo cuando esta copia normal restante se convierte mutado que la célula puede someterse al proceso de transformación maligna. El papel de los obstetras y ginecólogos implica:
Estratificación del riesgo: Riesgo esporádica, riesgo familiar y riesgo hereditarioMás del 10% de los pacientes tienen un historial de salud personal o familiar que sugiere la susceptibilidad al cáncer hereditario o familiar, y más del 6% de los pacientes que cumplen los criterios Red Nacional Integral Cáncer (NCCN) para las pruebas genéticas (3). Tres perfiles de riesgo han surgido:
Riesgo hereditario lleva el mayor porcentaje de susceptibilidad al cáncer, mientras que el riesgo esporádica conlleva el más bajo. Con la estratificación del riesgo, podemos identificar a las personas que pueden beneficiarse de la detección intensiva, pruebas genéticas y las intervenciones como la quimioprevención y la reducción del riesgo quirúrgico. Las pruebas genéticas de las personas adecuadas nos permite además identificar a los pacientes con síndromes de cáncer hereditario, para su propio beneficio y el de toda su familia. Una vez que se identifica una historia familiar de cáncer, los modelos se analizan a continuación se utilizan para predecir un cáncer particular y la probabilidad de una mutación genética que predispone al paciente a un síndrome de cáncer hereditario. El consentimiento informado, incluyendo los riesgos, beneficios, opciones y expectativas, se debe discutir adecuadamente. Asesoramiento directo es necesario en el caso de una familia anormal o cáncer de la historia personal. Asesoramiento para no directa reduce la oportunidad del paciente para aumentar la vigilancia y el diagnóstico precoz y la prevención potencial de cáncer, y el médico pone en riesgo de responsabilidad futura. Hereditario de mama y el síndrome de cáncer de ovario (HBOC)Mama hereditario y el cáncer de síndrome de ovario (HBOC) es un síndrome de cáncer hereditario de susceptibilidad. Las características de este síndrome son múltiples miembros de la familia con cáncer de mama o cáncer de ovario o de ambos, la presencia de cáncer de mama y cáncer de ovario en una sola persona y la edad temprana de aparición del cáncer de mama. A mediados de la década de 1990 se demostró que las mutaciones hereditarias en los genes BRCA1 y BRCA2 genes en los cromosomas 17 y 13, respectivamente, fueron los encargados de mama más familiar y cáncer de ovario. Tanto las mujeres como los hombres pueden llevar a un mutante BRCA gen y transmitirlo a su descendencia. "Banderas rojas" para el síndrome HBOC - antecedentes familiares y personales de tres generaciones incluyendo (4):
BRCA1 y BRCA2Aproximadamente el 10% de los casos de cáncer de ovario y el 3-5% de los casos de cáncer de mama se deben a mutaciones de línea germinal en los genes BRCA1 y BRCA2 (1),(5). BRCA1 se encuentra en el cromosoma 17, y BRCA2 está en el cromosoma 13. Más de 1.200 mutaciones diferentes han sido reportados para BRCA1 , y más de 1300 mutaciones diferentes han sido reportados para el BRCA2 . BRCA1 y BRCA2 son genes supresores de tumores que codifican proteínas que funcionan en el proceso de reparación del ADN (6). Aunque las personas con síndrome HBOC heredan un alelo defectuoso en BRCA1 y BRCA2 de su padre o de la madre, tienen un segundo alelo, funcional. Si el segundo alelo se vuelve no funcional, el cáncer se puede desarrollar a través de la acumulación de mutaciones adicionales. Esto se conoce como la "hipótesis de dos hit" (7). En población general, se estima que aproximadamente 1 en 300 a 1 en 800 individuos llevan una mutación en los genes BRCA1 y BRCA2 (8). La población judía Ashkenazi representa una notable excepción, ya que hay tres mutaciones fundadoras específicos (BRCA1 codón 185deletion AG, BRCA1 codón 5374 insert C y BRCA2 codón 6174 deleción T) que se realizan en cerca de 2,5% de los pacientes (8). En vista de ello, la mayoría de los expertos tienen un umbral mucho más bajo para las pruebas genéticas en las mujeres judías. Para una mujer, con un BRCA1 mutación, el riesgo de cáncer de ovario es 39-46%. Para una mujer con una BRCA2 mutación, el riesgo de cáncer de ovario es de 12-20%. El riesgo estimado de cáncer de mama con un BRCA1 o BRCA2 mutación es 65 a 70% (9). Para las mujeres con cáncer de mama, el riesgo actuarial de 10 años de desarrollar cáncer de ovario posterior es 12,7% para BRCA1 portadores de la mutación y el 6,8% para BRCA2 portadores de mutaciones (10). El cáncer de ovario asociado a BRCA1 y BRCA2 mutaciones tiene un fenotipo histológico distinto. Este tipo de cáncer es predominantemente de la histología serosa o endometrioide y es de alta calidad. Mucinosos y límite de los cánceres de ovario no parecen ser parte del espectro del tumor (11). Cáncer de las trompas de Falopio Primaria y el cáncer peritoneal primario también son del espectro de las enfermedades relacionadas con los genes BRCA1 y BRCA2 mutaciones (12). Criterios para la evaluación del riesgo genéticoLos pacientes con mayor que un 20-25% de probabilidad de tener una predisposición hereditaria al cáncer de mama y cáncer de ovario y para los cuales se recomienda la evaluación del riesgo genético (13):
*Cáncer de los tubos de Falopio peritoneo y debe ser considerada como una parte del espectro del síndrome HBOC. Cerrar relativa se define como un pariente de primer grado (madre, hermana, hija) o pariente de segundo grado (abuela, nieta, tía, sobrina). Los pacientes con más de un 5 a 10% de probabilidad de tener una predisposición hereditaria al cáncer de mama y cáncer de ovario y para los que la evaluación del riesgo genético puede ser útil (13):
Herramienta de puntuación del riesgo para el síndrome HBOCHemos desarrollado una herramienta de enseñanza numérica simple que se puede utilizar para estimar la candidatura de un paciente para BRCA pruebas. Esta herramienta se aproxima guías de la NCCN por asignar 1 o 2 puntos a cada persona con cada una "bandera roja" cáncer relevante en el árbol de familia de tres generaciones. Los puntos del paciente se añaden junto con los puntos maternas y luego de nuevo con los puntos paternos. Una suma de 0 puntos indicaría una clasificación esporádica (bajo). Una suma de 1 punto por lo general indica una clasificación de riesgo familiar (medio), sin embargo, un paciente de 1 punto todavía puede calificar para las pruebas genéticas si hay una estructura familiar limitada o una predisposición étnica de BRCA mutaciones. Una suma de> 2 puntos por lo general califican para BRCA pruebas, aunque las combinaciones que incluyen parientes de tercer grado pueden ser evaluados para determinar si la prueba se justifica. Esta herramienta sólo debe ser usado como una estimación, y no una guía de pruebas concluyentes. Cada comentario del Círculo aplicables a usted. Luego suma a + b, entonces a + c
Cada cáncer primario de cuenta por separado
HBOC, hereditary breast and ovarian cancer; LFS, Li-Fraumeni syndrome 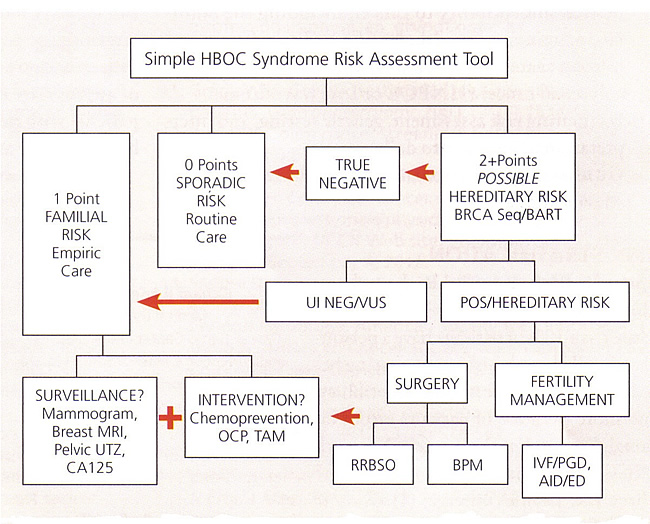 Abbreviations: AID, artificial insemination by donor; BART, BRCA Analysis rearrangement test; BPM, bilateral prophylactic mastectomy; CA 125, cancer antigen 125; ED, egg donation; HBOC, hereditary breast and ovarian cancer; IVF, in-vitro fertilization; MRI, magnetic resonance imaging; OCP, oral contraceptive pills; PGD, preimplantation genetic diagnosis; POS, positive; RRBSO, risk-reducing bilateral salpingo-oophorectomy; TAM, tamoxifen; UI NEG, uninformative negative; UTZ, ultrasound; VUS, variant of uncertain significance. Asesoramiento Genético para Reducir el Riesgo de Cáncer de MamaEl consejo genético debe incluir un análisis de los posibles resultados de las pruebas - abordar específicamente los temas de los resultados positivos, negativos y poco informativo, o variantes de significado desconocido. Opciones para la vigilancia, la quimioprevención y la cirugía de reducción del riesgo deben ser discutidos antes de la prueba. También se deben considerar las posibles implicaciones psicológicas y familiares de los resultados de las pruebas. Los materiales escritos pueden ayudar a las personas a compartir información con sus familiares sobre sus posibles riesgos genéticos. La sesión de asesoramiento genético también debe incluir una discusión sobre el costo de las pruebas genéticas. Muchas compañías de seguros, incluyendo Medicare, cubren una parte significativa de los gastos para ciertos individuos. Medicare y otras compañías de seguros han escrito guías para cubrir el costo de las pruebas genéticas. Un aspecto importante del asesoramiento genético es la discusión de la legislación vigente en materia de discriminación genética y la privacidad de la información genética. La Ley de Discriminación por Información Genética federal de 2008 protege a los individuos contra la salud y la discriminación en el empleo basada en la información genética. Muchos estados también tienen leyes estatales que proveen una protección similar. Estas leyes no se aplican a otros tipos de seguros, que pueden incluir seguros de vida o invalidez. Lo ideal es que tiene sentido para iniciar las pruebas en un individuo afectado. Si, sin embargo, ningún individuo afectado está disponible, las pruebas genéticas pueden todavía proporcionar información útil. Si se identifica una mutación deletérea, los pacientes en situación de riesgo pueden ser debidamente asesorados sobre las pruebas u otros métodos de reducción de riesgos. Si no se identifica una mutación deletérea, los pacientes deben ser aconsejados que esto podría ser debido a una de las varias posibilidades que incluye (a) una deletérea mutación presente en la familia que el paciente no heredó, (b) una mutación detectable en los genes BRCA1 y BRCA2 , o un gen de susceptibilidad al cáncer sin identificar todavía está presente en la familia, y no se sabe si el paciente comparten esta predisposición, o (c) hay predisposición hereditaria en la familia. Las estrategias actuales para reducir el riesgo de desarrollar cáncer de ovario o cáncer de las trompas de Falopio en las mujeres con alto riesgo con nocivo conocido BRCA mutaciones incluyen la vigilancia, la quimioprevención y la cirugía. Teniendo en cuenta el elevado riesgo de cáncer de ovario y cáncer de las trompas de Falopio en las mujeres con mutaciones en los genes BRCA1 y BRCA2 , grupos de consenso han recomendado la revisión periódica con CA 125 y la ecografía transvaginal, comenzando entre las edades de 30 años y 35 años o 5-10 años antes que la edad más temprana del primer diagnóstico de cáncer de ovario en la familia (14). La baja prevalencia de cáncer de ovario y la alta probabilidad de un resultado de la prueba de detección positiva que requiere mayor evaluación quirúrgica invasiva son obstáculos en los programas de cribado de cáncer de ovario entre las mujeres en riesgo hereditario (15). Se espera que en el futuro juicio cáncer de ovario se beneficiará de mejores marcadores séricos y la mejora de los algoritmos de detección para mejorar la capacidad de discriminar entre 125 valores de CA normales y anormales y los hallazgos ecográficos. Los beneficios y la magnitud de la reducción del riesgo con los anticonceptivos orales para mujeres con BRCA mutación no se ha informado de la manera más coherente para la población general y de bajo riesgo. Es razonable que las mujeres con mutaciones en los genes BRCA1 y BRCA2 , que el uso de anticonceptivos orales. La mayoría de los estudios reportan un menor riesgo de cáncer de ovario entre los que usaron anticonceptivos orales durante más tiempo (más de 3 a 6 años) (16). Los riesgos relativos y los beneficios, tanto para la quimioprevención y control reproductivo deben sopesarse cuidadosamente por el paciente y su médico. Profiláctica salpingo-ooforectomía bilateral (BSO)Se recomienda encarecidamente a las mujeres que llevan BRCA mutaciones debido a la alta tasa de mortalidad de cáncer de ovario y la falta de investigación efectiva y métodos de prevención (17). Afortunadamente, el riesgo de cáncer de mama y ovario hereditario y no aumentar de manera espectacular hasta finales de los años 30 en las mujeres con BRCA1 mutaciones y finales de los 50 para las mujeres con BRCA2 mutaciones (18), por lo que las mujeres tienen la oportunidad de completar sus familias antes de someterse BSO. La práctica anterior de realizar la cirugía profiláctica basada únicamente en los antecedentes familiares en gran medida se debe abandonar. La mitad de las mujeres en las familias con BRCA no se espera que las mutaciones pueden ser portadores. Actualmente, la decisión de proceder con la cirugía BSO profiláctico se basa principalmente en los resultados de BRCA análisis mutacional. Existe alguna evidencia de que ciertos tipos de BRCA mutaciones pueden predisponer más fuertemente al cáncer de ovario, pero esta observación no ha sido confirmada consistentemente (19). Aunque BSO es un procedimiento quirúrgico mayor, los estudios muestran que la mayoría de las mujeres de alto riesgo sometidos a pruebas genéticas aceptan salpingo-ooforectomía bilateral. La laparoscopia y la laparotomía son a la vez las opciones de reducción de riesgos BSO. En ambos procedimientos, es necesaria una inspección a fondo de las superficies peritoneales. Lavados peritoneales deben obtenerse. El diafragma, hígado, epiplón, intestino, canalones paracólicas, y el apéndice son inspeccionados en el abdomen. Los ovarios, las trompas de Falopio, el útero, la vejiga serosa y cul-de-sac se inspeccionan en la pelvis. Las áreas anormales deben someterse a biopsia. Los vasos ováricos deben ser aislados y se ligaron proximal al extremo de tejido ovárico de identificación para asegurar que todo el tejido ovárico se elimina por completo. Si no se está realizando una histerectomía, la trompa de Falopio se debe dividir en su inserción en el cuerno uterino. Cuando se realiza un procedimiento laparoscópico, para optimizar la conservación de la superficie del epitelio ovárico, los especímenes pueden ser colocados en una bolsa antes de la extracción endoscópica de abdomen. La decisión de realizar una histerectomía concurrente debe ser individualizada. Los argumentos a favor de la histerectomía son una estrategia de terapia hormonal más simple (con sólo estrógeno) y un aumento en el riesgo teórico de cáncer en la trompa de Falopio corneal (20). Además, la histerectomía puede ser considerado cuando hay otras indicaciones médicas para la extirpación del útero y cuello uterino. Para las mujeres que toman tamoxifeno, la histerectomía se puede considerar para reducir su riesgo de cáncer de endometrio (21). Pros y Contras de Riesgo Reduciendo Ooforectomía Salpingo
Contras:
¿A qué edad se debe considerar la reducción de riesgos-ooforectomía Salpingo?De reducción de riesgos y electivos salpingo-ooforectomías son la eliminación de los ovarios y las trompas de Falopio para que el beneficio potencial de la prevención de la morbilidad y la mortalidad a largo plazo. El plazo de reducción de riesgos salpingooforectomía implica que los ovarios son normales en el momento de la extracción. Las mujeres con BRCA1 o BRCA2 mutaciones se les debe ofrecer la reducción de riesgos salpingooforectomía por la edad de 40 años, o cuando se haya completado la maternidad (13). El cáncer de ovario se diagnostica en menos de un 2-3% de las mujeres con BRCA1 o BRCA2 mutaciones antes de los 40 años. Para las mujeres con BRCA1 mutaciones, el riesgo de cáncer de ovario aumenta notablemente durante los años 40, con 10-21% de BRCA1 portadores de mutaciones en desarrollo el cáncer de ovario por la edad de 50 años. El riesgo de cáncer de ovario antes de la menopausia es mucho menor en BRCA2 portadores de mutaciones, con no más de 3% de BRCA2 portadores de mutaciones en desarrollo el cáncer de ovario por la edad de 50 años (13),(22). Dado el diferente calendario de riesgo de cáncer de ovario, el examen se puede hacer para aconsejar pacientes con BRCA1 mutaciones diferentes que para BRCA2 portadores de mutaciones. Sin embargo, las mujeres con BRCA2 mutaciones tienen una probabilidad 26-34% de desarrollar cáncer de mama en 50 años (18),(22), y el máximo beneficio de la eliminación de los ovarios en la reducción del riesgo de cáncer de mama se consigue cuanto antes se extirpan los ovarios (23). Teniendo en cuenta estas cuestiones, el calendario de reducción del riesgo salpingo-ooforectomía bilateral debe basarse en las necesidades individuales de cada paciente, teniendo en cuenta su deseo de preservar la fertilidad o prevenir prematura menopausia quirúrgica con el impacto depende de la edad de la reducción del riesgo salpingo-ooforectomía bilateral, tanto de mama cáncer y los riesgos de cáncer ginecológico. Salpingectomía Profiláctica. Ooforectomía RetardadaProfiláctica salpingo-ooforectomía bilateral se recomienda para las mujeres con BRCA mutaciones, pero hay consecuencias adversas de la menopausia prematura. La mayoría de BRCA cánceres ováricos asociados parecen surgir en la trompa de Falopio, por lo tanto, salpingectomía puede ser una alternativa a la salpingooforectomía bilateral. Con el entendimiento de que tal vez 60% de los cánceres pélvicos serosas puede originarse en la trompa de Falopio, ha habido una discusión más reciente de reducción del riesgo salpingectomía como una alternativa potencial a la reducción del riesgo BSO para el tratamiento de BRCA portadores de la mutación, en particular en las mujeres reacios a someterse a reducción del riesgo BSO (39). Los autores publican sus Markov Monte Carlo de simulación comparando BSO de reducción del riesgo a los 40 años de edad con tanto salpingectomía de reducción del riesgo a los 40 años de edad, seguido de una ooforectomía bilateral retraso a los 50 años de edad (40). Este modelo reflexivo predice la reducción esperada en los cánceres pélvicos serosos, los cánceres de mama y muertes adicionales debidas a la enfermedad cardiovascular. La conclusión del estudio fue salpingooforectomía bilateral a menudo la mayor reducción del riesgo de cáncer de ovario y de mama entre las portadoras de mutaciones BRCA. Sin embargo, cuando se considera la esperanza de vida ajustada a la calidad, salpingectomía bilateral con ooforectomía tardía es una estrategia rentable y puede ser una alternativa aceptable para aquellos que no quieren someterse a salpingo-ooforectomía bilateral (40). Es importante destacar que el estándar de atención para las mujeres que heredan mutaciones germinales en los genes BRCA1 y BRCA2 sigue siendo BSO profiláctico después de la finalización de la maternidad o alrededor de la edad de 40 años. Ofrece la mayor reducción del riesgo en el cáncer de mama y de ovario en comparación con salpingectomía con o sin ooforectomía retardada. Sin embargo, una proporción significativa de mujeres no se someten a BSO, y muchos eligen vigilancia sola para el cáncer de ovario a pesar de la limitada beneficio de los métodos de detección existentes (41). El cáncer de ovario impulsa la tasa de mortalidad entre BRCA portadores de la mutación, por lo tanto, la intervención que reduce el riesgo de cáncer de ovario es probable que sea validado prospectivamente, salpingectomía bilateral con ooforectomía retraso puede ser una alternativa razonable a la BSO, especialmente para aquellos que son reacios a someterse a este último procedimiento debido al efecto potencial en la calidad de vida. Gestión de las mujeres con antecedentes familiares fuertes y negativas BRCA o BRCA2 mutaciónAunque, en la mayoría de los casos, una predisposición hereditaria al cáncer de ovario es causada por mutaciones en BRCA1 o BRCA2 , la tecnología actual no permite la identificación de todas las mutaciones que deben existir en estos genes (24). Además, estudios de ligamiento han sugerido que es menos de la mitad de las familias con cuatro o más casos de cáncer de mama, pero no hay casos de cáncer de ovario (familias con cáncer de mama de sitio específico), cáncer de mama son causados por genes BRCA1 o BRCA2 mutación (25). Teniendo en cuenta estas cuestiones, las mujeres con una historia personal o familiar de cáncer de mama que han dado negativo para un BRCA mutación deben gestionarse en base a su historia familiar. Los datos preliminares sugieren que las mujeres de familias con antecedentes de cáncer de mama de sitio específico en el que no BRCA se identifica la mutación se mantienen en un aumento significativo del riesgo de cáncer de mama, pero no pueden estar en un riesgo significativamente mayor de cáncer de ovario (26). Es importante para las personas de alto riesgo a mantenerse en contacto con los médicos con experiencia en el cuidado de las mujeres con mayor riesgo, dada rápido desarrollo de la investigación y las mejoras en la tecnología de pruebas. Por ejemplo, una prueba de grandes reordenamientos en el gen BRCA1 y BRCA2 genes se ha desarrollado que puede ayudar a identificar las mutaciones en un pequeño porcentaje de las familias de alto riesgo que previamente arrojaron resultados negativos para estos genes. Opciones de reproducciónLos resultados de las pruebas genéticas pueden tener un profundo impacto en las decisiones de planificación familiar para las personas en edad reproductiva que se encuentran para ser portadores de BRCA1 / 2 mutaciones. Por ejemplo, en los casos en que ambos socios llevan un BRCA2 mutación, puede haber un alto riesgo para la descendencia de desarrollar una rara anemia de Fanconi / fenotipo de tumor cerebral (trastorno recesivo). Consejería de opciones reproductivas como el diagnóstico prenatal, el diagnóstico genético preimplantacional (DGP) y la reproducción asistida por tanto, puede estar justificado para parejas que expresan su preocupación por el BRCA estado de portador de la mutación de su futura descendencia. Este asesoramiento debería incluir una discusión completa de los posibles riesgos, beneficios y limitaciones de las opciones reproductivas. El diagnóstico prenatal implica la post-implantación análisis genético de un embrión temprano, la utilización de las vellosidades coriónicas o muestras de células del líquido amniótico, las pruebas genéticas se suele realizar entre la semana 12 y la semana 16 de gestación, y los resultados de pruebas puede dar lugar a la decisión de una pareja para interrumpir el embarazo (27). Durante las últimas dos décadas, el DGP se ha convertido en un método alternativo de las pruebas genéticas en embriones tempranos. PGD implica la prueba de 1 o 2 células de embriones en fases muy tempranas de desarrollo (es decir, de 6 a 8 células) después de la fertilización in vitro (FIV). Este procedimiento permite la selección de embriones no afectados para ser transferidos al útero, y puede, por lo tanto, ofrecen la ventaja de evitar la potencial interrupción del embarazo. Sin embargo, los procedimientos tales como PGD no carecen de limitaciones, ya que podría solicitar un diagnóstico prenatal de confirmación en función de las necesidades o solicitudes de médicos de la pareja. Por otra parte, el proceso de PGD requiere el uso de la FIV sin importar el estado de fertilidad de la pareja (es decir, se aplica también a las parejas sin problemas de infertilidad), y la FIV no siempre puede dar lugar a un embarazo exitoso. Por último, la tecnología o la experiencia pueden no estar disponibles en la ubicación geográfica de la pareja. Varios factores, tanto médicos como personal, se deben pesar en la decisión de utilizar el diagnóstico prenatal o PGD. Consideraciones médicas pueden incluir factores tales como la edad de inicio del cáncer, penetrancia, la gravedad o la morbimortalidad del cáncer hereditario, y la disponibilidad de los métodos de reducción de riesgo de cáncer eficaces o tratamientos eficaces. Aunque el uso de diagnóstico prenatal o PGD está relativamente bien establecido para los trastornos hereditarios graves con muy alta penetrancia, su uso en condiciones asociadas con penetrancia inferior (por ejemplo, de mama hereditario o síndrome de cáncer de ovario) sigue siendo algo controvertido, tanto desde el punto de vista ético y normativo. Consideraciones personales para la decisión de utilizar el diagnóstico prenatal o PGD pueden incluir creencias éticas individuales, sistemas de valores, las creencias culturales y religiosas, así como los factores sociales y económicos. Con base en los resultados de encuestas realizadas a las mujeres con alto riesgo de cáncer de mama o de ovario hereditario, 50% -75% de los encuestados consideró que el DGP es una opción aceptable para el individuo de alto riesgo sin embargo, sólo alrededor del 14% -33% consideraría someterse a PGD mismos (28). Es importante destacar que las encuestas sugieren que la mayoría de las mujeres de alto riesgo tienen poco o ningún conocimiento de PGD, destacando la necesidad de una mayor sensibilización y educación acerca de las posibles opciones reproductivas. Nacimientos exitosos han sido reportados con el uso de la PGD e IVF en BRCA1 / 2 portadores de la mutación, pero los datos disponibles en la literatura publicada aún muy limitada. Además, los datos relativos a la seguridad o los resultados de la reproducción asistida en PDG y largo plazo BRCA portadores de la mutación aún no están disponibles. Poliposis hereditario del cáncer de colon (CCHNP) o síndrome de LynchEs una enfermedad autosómica dominante heredado del sistema de reparación de ADN no coinciden. Muchas autoridades se refieren a HNPCC como el síndrome de Lynch, creyendo que HNPCC no tiene en cuenta adecuadamente la importancia de malignidades extracolónicas, tales como endometrio, de ovario, tracto gastrointestinal superior, y cánceres del tracto urinario asociados con esta enfermedad. Este síndrome representa el 5-10% de todos los cánceres de colon y de endometrio (29). El análisis de ligamiento de las familias de alto riesgo llevó al descubrimiento de que el síndrome de Lynch es causado por mutaciones en la línea germinal en una clase de genes responsables de la reparación de ciertos tipos de mutaciones del ADN (29). Estos genes se denominan "desajuste reparación de" genes y dan lugar a proteínas que corregir ADN y corregir los errores que se realizan durante el proceso normal de la replicación. MSH2 (homólogo de MutS 2) y MLH1 (homólogo 1 de MutL L) son la falta de coincidencia más comúnmente mutado reparar genes en este síndrome. MSH2 y MLH1 se encuentran en los cromosomas 2p16 y 3p21, respectivamente. Mutaciones de la línea germinal en otros genes de reparación ( MSH6 , PMS1 y PMS2 ) han sido identificados, pero a una frecuencia más baja (29). Probabilidad de desarrollar un cáncer si hay línea germinal MLH1 o MSH2 mutaciones:
La historia familiar es el primer paso para determinar si un paciente está en mayor riesgo de una enfermedad hereditaria. Varios criterios relativamente estrictas se han desarrollado para identificar a los individuos con alto riesgo de síndrome de Lynch. Alrededor del 30% de las familias que cumplen los criterios de Bethesda y de 50 a 92% de familias que cumplen los criterios de Amsterdam tendrán desajuste mutación de la línea germinal de genes de reparación del DNA (30),(31). Los criterios de Bethesda para CCHNP (ver más abajo) parecen ser los más sensibles en la predicción de mutaciones de reparación de desajuste en las familias HNPC (30). Cualquiera de los siguientes:
Los criterios de Ámsterdam II (ver más abajo) del HNPCC son más específicos (31). Todos los siguientes:
Herramienta de Evaluación de Riesgo para HNPCC / Síndrome de Lynch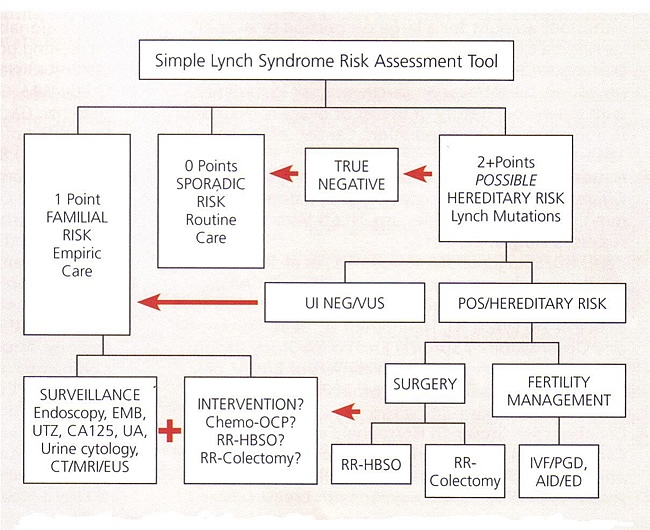 Abbreviations: AID, artificial insemination by donor; CA 125, cancer antigen 125; CT, computed tomography, ED, egg donation; EMB, endometrial biopsy; EUS, endoscopic ultrasound; HBSO, hysterectomy with bilateral salpingo-oophorectomy; IVF, in-vitro fertilization; MRI, magnetic resonance imaging; OCP, oral contraceptive pills; PGD, preimplantation genetic diagnosis; POS, positive; RR, risk-reducing; UA, urinalysis; UI NEG, uninformative negative; UTZ, ultrasound; VUS, variant of uncertain significance. La vigilancia de los cánceres de colon sin poliposis hereditario (HNPCC)Cuando se identifican las mutaciones, un menos costoso análisis genético simple,, se puede realizar en otros miembros de la familia. Asesoramiento extenso se recomienda antes y después de la prueba. Una vez que el proceso genético ha sido completado, el riesgo de cáncer puede ser asignado para el paciente y su familia. Cuando una mutación ha sido identificada, la prueba genética se debe ofrecer a todos los miembros de la familia. Estas personas deben someterse a cada consejo genético por lo que las ventajas y desventajas de las pruebas pueden ser explicadas. Los portadores de una mutación se ofrecen vigilancia intensiva. Si no hay mutación se identificó en el paciente, pero la familia se reúne criterios de Amsterdam el protocolo para el síndrome de Lynch, la vigilancia debe observar. La mortalidad por cáncer colorrectal en este grupo ha sido demostrado ser reducido por la investigación de la colonoscopia (32). La mayoría de las autoridades creen que el riesgo de reducción de la histerectomía con salpingooforectomía bilateral tiene un papel en el manejo de estos pacientes. Los cánceres colorrectales en el síndrome de HNPCC tienen una propensión a ocurrir en el colon proximal, en relación con los casos esporádicos, que son a menudo más distal. Colectomía profiláctica se recomienda a los pacientes con mutación HNPCC en el que se identifican los pólipos a una edad temprana, y también a los pacientes que tienen pólipos que son la inestabilidad de microsatélites-positivo o aquellas personas que no pueden someterse a la vigilancia regular. La quimioprevención con agentes antiinflamatorios no esteroides en este grupo de pacientes para prevenir los pólipos de colon se encuentra actualmente en estudio, y los datos debe ser inminente. Si se están estudiando estos pacientes por otra razón, debe considerarse la posibilidad de asesoramiento en relación con los posibles riesgos y beneficios de una histerectomía total con salpingooforectomía bilateral. La vigilancia de HNPCC (30),(33):
GI, gastrointestinal Síndrome de Li-FraumeniSíndrome de Li-Fraumeni (LFS) se asocia con mutaciones de línea germinal en el gen supresor de tumores TP53 , que causan una predisposición muy penetrantes para desarrollar sarcomas, así como de mama de aparición temprana, el cerebro, la corteza suprarrenal, y otros tipos de cáncer. Las personas con LFS tienen un riesgo absoluto de desarrollar cualquier tipo de cáncer de aproximadamente 50% en los 30 años y el 90% antes de los 60 años (34). LFS cuenta para un máximo de 1% de cáncer de mama en Estados Unidos; tales cánceres pueden tener un fenotipo distintivo, que expresan receptores de hormonas y HER2/neu. La frecuencia de mutación de novo se estima en 7% a 20%, de tal manera que TP53 pruebas puede ser apropiado para pacientes seleccionados en la ausencia de una historia familiar de cáncer (34). Síndrome de CowdenEl síndrome de Cowden (CS) transmite una mayor susceptibilidad a la mama, tiroides, endometrio y colon y hamartomas benignos que afectan a múltiples órganos. Es un trastorno autosómico dominante asociado con mutaciones en el gen supresor tumoral PTEN . Tumores malignos ginecológicos son comunes con un riesgo 5-10% de cáncer de endometrio y 25-50% de riesgo de cáncer de mama (42). Manifestaciones externas incluyen macrocefalia y papilomatosas pápulas en la cara, mucosa oral-lingual, y las extremidades, que son casi omnipresentes en la edad de 30 años. El riesgo de cáncer de mama por vida es de 25-50%, con una edad media de diagnóstico de 38 a 46 años (35). El riesgo de por vida de cáncer de endometrio es de 5-10% (35). Mutación germinal en el PTEN gen se identifican en aproximadamente el 80% de los pacientes que cumplían los criterios diagnósticos clínicos, casi la mitad de los cuales no tienen antecedentes familiares de CS. La frecuencia informado de CS es 1 en 200.000, pero es probable infradiagnosticada (35). Recurrentes, múltiples pólipos endometriales auguran un alto riesgo de cáncer de endometrio en las mujeres con CS. Monitoreo de malignidad y la consideración de la histerectomía después de la maternidad se ha completado se justifica (42). Síndrome de cáncer gástrico difuso hereditarioSíndrome de cáncer gástrico difuso hereditario se asocia con mutaciones en el gen CDH1 gen y se caracteriza por un riesgo de por vida 80% de desarrollar cáncer gástrico (36). Las mujeres con CDH1 mutaciones tienen hasta un riesgo de por vida del 60% de desarrollar cáncer de mama lobular, sin embargo, la prevalencia de CDH1 mutaciones en mujeres seleccionadas para la edad temprana de inicio de síntomas o antecedentes familiares de cáncer de mama lobular es baja, alrededor del 1% (36). Entre las familias con cáncer gástrico difuso y lobulillar de mama, CDH1 mutaciones se identifican en el 25-50% (36). Síndrome de Peutz-JeghersEl síndrome de Peutz-Jeghers (PJS), dando como resultado de alteraciones en la STK11 gen, cuenta con pólipos hamartomatosas en todo el tracto gastrointestinal, lo que lleva a la invaginación intestinal y obstrucción intestinal. STK11 1 mutaciones transmiten un mayor riesgo de cáncer de mama, de ovario, de cuello uterino, de páncreas, gástrico, y de colon cáncer, además de los tumores del cordón sexual benignos. El riesgo de cáncer de mama alcanza el 8% en los 40 años y 32% a la edad de 60 años. La mitad de STK11 portadores de la mutación carecen de antecedentes familiares de PJS (37). La neurofibromatosis-1La neurofibromatosis-1 (NF-1) Características neurofibromas cutáneos y los tumores de la vaina nerviosa periférica tumores, problemas de aprendizaje, y una prevalencia de 1 en 3.000 nacidos vivos (38). Un estudio reciente de mujeres con NF-1 reportó un aumento cinco veces el riesgo de desarrollar cáncer de mama antes de los 50 años y un 3 - a 4 veces mayor en el riesgo de cáncer de toda la vida (38). Fomento de la Calidad de la atención del cáncerComo la detección de cánceres hereditarios se ha vuelto más fácilmente disponible, muchas preguntas en torno a la responsabilidad, han surgido gestión de riesgos y seguridad del paciente. Al igual que en todas las cuestiones médico-legales, estas áreas de interés general, se refieren a la norma de la atención, la documentación, el consentimiento, las expectativas de los pacientes y el seguimiento. Muchos profesionales consideran que la detección del cáncer hereditario no es estándar de la atención en el consultorio de atención primaria. Sin embargo, hay tres puntos que son muy importantes para recordar. Es norma de atención para obtener una amplia y completa historia de la familia y actualizarlo de forma rutinaria. Es norma de atención para dar a los pacientes la información adecuada sobre la base de que la historia de la familia para que puedan tomar decisiones informadas acerca de su atención médica. Por último, es norma de atención a fondo y completamente grabar lo que se discutió con el paciente. Si se adhieren a estos tres puntos, entonces parecería que la detección del cáncer hereditario es, de hecho, el nivel de atención. Documentación: una vez que se ha identificado a alguien que se ajuste a los criterios de las pruebas genéticas, la cantidad de documentación que se necesita? ¿Es adecuado para que su estado de cuenta: "Información sobre las pruebas genéticas dado" o "folleto da?" aunque es agradable ver su plan documentado, es mucho más importante ver el razonamiento detrás del plan. En este caso, una nota expandida, tales como:. ", Basada en la historia familiar, las pruebas genéticas recomendado paciente entiende que si la prueba es positiva hay un aumento sustancial en el riesgo de cáncer de ovario y / o cáncer de mama o [el cáncer de síndrome de Lynch en particular que el tamizaje] ". Aunque sabemos que hablamos de los riesgos de cáncer, el paciente puede estar en contradicción con facilidad lo que no está documentado en su carta. Los pacientes pueden argumentar que si entendieron sus riesgos, ellos, por supuesto, han dado su consentimiento para la prueba. La incorporación de algún tipo de sistema de seguimiento en su oficina es prudente. Esto puede permitir que el seguimiento de un paciente después de que se ha remitido para el consejo genético. Sin este tipo de rastreo y seguimiento, una cuestión preocupante puede ser elevado: "si sentía que era lo suficientemente importante como para que el paciente tiene esta prueba, ¿por qué no era lo suficientemente importante para que usted pueda ver si la prueba se llevó a cabo" Consentimiento Informado: El consentimiento informado o el rechazo informado deben abordarse al examinar las evaluaciones de riesgo hereditarios. Por lo general, el consentimiento informado ha tratado solamente con informar a los pacientes de los riesgos asociados a procedimientos invasivos. Sin embargo, ha habido una expansión de lo que incluye el consentimiento informado adecuado. Como parte del consentimiento informado adecuado que ahora se pide para dar todas las opciones de tratamiento, así como los riesgos y beneficios de cada opción. Por lo tanto, si no nos damos por pacientes apropiados la opción de las pruebas genéticas (junto con sus riesgos y beneficios), se podrá considerar que una negligencia en una base consentimiento en caso de haber un evento adverso. Aquí es donde rechazo informado puede entrar en juego, si un paciente no quiere hacer lo que el proveedor se siente es apropiado, o no se ha seguido con una referencia consejo genético cuando la envió a uno, la documentación de su negativa, o la falta de seguimiento en marcha, en última instancia puede ser más importante que la documentación de su consentimiento. Documentos rechazo informado de que el médico haya hecho lo que es prudente y que es elegido por el paciente para no seguir adelante. Muchos estados tienen algún elemento de negligencia, y esto puede dar un paso más y documentar el motivo de la negativa del paciente, el miedo del resultado de la prueba, falta de voluntad para hacer nada sobre el resultado, o por razones financieras puede ser parte de la decisión de un paciente rechazar la prueba. En la actualidad, una de las principales causas de los casos de negligencia implica problemas con el cáncer de mama. Por lo general, las denuncias incluyen tanto el diagnóstico como la falta de diagnóstico tardío. Ahora estamos viendo una nueva alegación que se conoce como un fracaso de nuestro "deber de informar" o "deber de advertir". Se podrían haber realizado Esto se refiere a la falta de identificación de un paciente en riesgo de un cáncer hereditario por lo que el aumento de la vigilancia podría haber sido aplicado para diagnosticar el cáncer o antes que la cirugía de reducción del riesgo o profilácticos. Este tipo de casos va a ser muy difícil, si no imposible, para defender sin la documentación adecuada, incluyendo la documentación de la negativa de las pruebas de un paciente, y la documentación de la explicación de los riesgos muy específicos de cáncer. ResumenMás de 300.000 mujeres en Estados Unidos se estima que llevar a un alto riesgo hereditario de padecer cáncer de mama y de ovario. Una vez más amplia gama de pruebas genéticas está disponible para definir el riesgo del paciente, sin embargo, los resultados aún puede resultar concluyentes o complicado de interpretar. Profesionales de la genética del cáncer ofrecen amplia determinación del riesgo, asesoramiento y recomendaciones para la gestión de los pacientes y las familias con una predisposición hereditaria a desarrollar cáncer de mama y de ovario. Como especialistas en salud de la mujer, la evaluación del riesgo hereditario es nuestra responsabilidad. Los antecedentes familiares de cáncer centrado debe ser parte de la evaluación inicial de los pacientes. Pedigree materno y paterno se deben construir para incluir por lo menos tres generaciones (del paciente, sus padres y sus abuelos generaciones). La información relativa a la edad del miembro de la familia al momento del diagnóstico y los detalles adicionales, tales como el cáncer de mama en los dos pechos son muy serviciales. Actualizaciones de la historia de la familia se les debe preguntar acerca de los exámenes anuales ya las adiciones y nuevos diagnósticos pueden alterar la recomendación inicial sobre las pruebas genéticas. Se recomienda una evaluación del riesgo genético para los pacientes con un mayor que un 20-25% de probabilidad de tener una predisposición hereditaria al cáncer de mama y cáncer de ovario. Las mujeres con BRCA1 o BRCA2 mutaciones se les debe ofrecer la reducción de riesgos salpingooforectomía de 40 años de edad o cuando la maternidad se ha completado. Para obtener una reducción del riesgo salpingooforectomía bilateral, todo el tejido de los ovarios y las trompas de Falopio se debe quitar. Visualización minuciosa de las superficies peritoneales con lavados pélvicos se debe realizar. Seccionamiento completo, serial de los ovarios y las trompas de Falopio es necesario, con el examen microscópico de cáncer oculto. Referencias
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||