Hereditary Cancer Risk Assessment in GynecologyWHEC Practice Bulletin and Clinical Management Guidelines for healthcare providers. Educational grant provided by Women's Health and Education Center (WHEC). Hereditary cancer risk assessment should be part of routine obstetrics and gynecology practice. Over the last decade there has been an increased awareness of hereditary predisposition to a wide range of diseases including cancer, heart diseases, and diabetes. These are all complex multi-gene disorders, but the identification of specific genes associated with a predisposition to these conditions has allowed clinicians to more accurately assess risk and prescribe preventive interventions. Though it may be unfamiliar to many practitioners, the process of cancer risk stratification can be efficient and effective. Using protocol-driven evaluation of cancer susceptibility, personal and family risk factors, and genetic testing, it is possible to create risk profiles and management strategies that demonstrate proven reduction in cancer morbidity and mortality. Decades before genetic testing entered practice, clinicians had an undue burden of breast and ovarian cancer, often featuring early-onset cases and male breast cancer. In the mid-1990s linkage analysis in these breast and ovarian cancer families enabled the identification of BRCA1 and BRCA2, genes that normally function in DNA repair. Cancer genetic counselors use pedigree analysis and clinical indicators to assess the probability that a patient carries a heritable mutation, to determine appropriated testing strategies among an increasingly complex array of commercially available assays, and to interpret results and guide medical recommendations. The scientific discovery, widespread public awareness and the general availability of clinical genetic testing of the BRCA1 and BRCA2 genes responsible for majority of familial breast and ovarian cancers, have led women to present to physicians offices requesting genetic testing and questioning the clinical implications for them and their families. The purpose of this document to review current recommendations for genetic testing for susceptibility to cancers, including ovarian, fallopian tube, breast, endometrial, and colon cancers due to inherited mutations in the BRCA genes or in the mismatch repair genes associated with hereditary nonpolyposis colon cancer (HNPCC) syndrome. Family history remains the cornerstone of patient identification. Ethnicity and race are routinely evaluated as a part of the assessment. Practical applications of genetic testing for cancer susceptibility have the ability to reduce the burden of hereditary cancers by saving lives, decreasing medical morbidities, and reducing psychological stress. Management of mutation carriers, including indications for risk-reducing surgeries, cancer screening, and follow-up, are also addressed. The review also focuses on clinical practices and the practical use of clinical cancer genetics. BackgroundThere are numerous syndromes that confer and inherited risk of breast, ovarian, and related cancers with 5% to 15% of all breast and ovarian cancer caused by one of these autosomal, dominantly inherited cancer syndromes (1),(2). The genes associated with hereditary cancer syndromes are all transmitted in a Mendelian autosomal dominant fashion, so maternal and paternal family histories contribute equally to the patients risk and need to be evaluated to determine the appropriateness of testing. It should be noted, however, that, although the gene mutations are inherited in an autosomal dominant fashion, they are expressed in a recessive manner (2). An individual who inherits a germline mutation that inactivates one copy of the gene in general has a second functionally intact copy of the gene. It is only when this remaining normal copy becomes mutated that the cell may undergo the process of malignant transformation. The role of the obstetricians and gynecologists involves:
Risk Stratification: Sporadic Risk, Familial Risk, and Hereditary RiskMore than 10% of patients have a personal or family health history suggesting hereditary or familial cancer susceptibility, and more than 6% of patients meet National Comprehensive Cancer Network (NCCN) criteria for genetic testing (3). Three risk profiles have emerged:
Hereditary risk carries the highest percentage of cancer susceptibility, while sporadic risk carries the lowest. With risk stratification, we can identify individuals who may benefit from intensive screening, genetic testing, and interventions such as chemoprevention and surgical risk reduction. Genetic testing of appropriate individuals further enables us to identify patients with hereditary cancer syndromes, for their own benefit as well that of their entire family. Once a family history of cancer is identified, models discussed below are used to predict a particular cancer and the likelihood of a genetic mutation that predisposes the patient to a hereditary cancer syndrome. Informed consent, including risks, benefits, options, and expectations, should be adequately discussed. Direct advice is necessary in the case of an abnormal family or personal cancer history. Non-direct counseling reduces the patients opportunity for increased surveillance and potential early diagnosis and prevention of cancer, and puts the physician at risk for future liability. Hereditary Breast and Ovarian Cancer (HBOC) SyndromeHereditary breast and ovarian cancer (HBOC) syndrome is an inherited cancer-susceptibility syndrome. The hallmarks of this syndrome are multiple family members with breast cancer or ovarian cancer or both, the presence of both breast cancer and ovarian cancer in a single individual and early age of breast cancer onset. In the mid-1990s it was demonstrated that inherited mutations in the BRCA1 and BRCA2 genes, on chromosomes 17 and 13 respectively, were responsible for most familial breast and ovarian cancers. Both women and men may carry a mutant BRCA gene and pass it on to their offspring. "Red Flags" for HBOC syndrome Personal and three-generation family history including (4):
BRCA1 and BRCA2Approximately 10% of cases of ovarian cancer and 3-5% of cases of breast cancer are due to germline mutations in BRCA1 and BRCA2 (1),(5). BRCA1 is found on chromosome 17, and BRCA2 is on chromosome 13. More than 1,200 different mutations have been reported for BRCA1, and more than 1,300 different mutations have been reported for BRCA2. BRCA1 and BRCA2 are tumor suppressor genes that encode proteins that function in the DNA repair process (6). Although individuals with HBOC syndrome inherit one defective allele in BRCA1 and BRCA2 from their father or mother, they have a second, functional allele. If the second allele becomes non-functional, cancer can develop through the accumulation of additional mutations. This is called the "two-hit hypothesis" (7). In general population, it is estimated that approximately 1 in 300 to 1 in 800 individuals carry a mutation in BRCA1 and BRCA2 (8). The Ashkenazi Jewish population represents a notable exception because there are three specific founder mutations (BRCA1 codon 185deletion AG, BRCA1 codon 5374 insert C, and BRCA2 codon 6174 deletion T) that are carried by about 2.5% of individuals (8). In view of this, most experts have a much lower threshold for genetic testing in Jewish women. For a woman, with a BRCA1 mutation, the risk of ovarian cancer is 39-46%. For a woman with a BRCA2 mutation, the risk of ovarian cancer is 12-20%. The estimated lifetime risk of breast cancer with a BRCA1 or BRCA2 mutation is 65-70% (9). For women with breast cancer, the 10-year actuarial risk of developing subsequent ovarian cancer is 12.7% for BRCA1 mutation carriers and 6.8% for BRCA2 mutation carriers (10). Ovarian cancer associated with BRCA1 and BRCA2 mutations has a distinct histologic phenotype. This type of cancer is predominantly of serous or endometrioid histology and is high grade. Mucinous and borderline ovarian cancers do not appear to be part of the tumor spectrum (11). Primary fallopian tube cancer and primary peritoneal cancer also are of the spectrum of disease associated with BRCA1 and BRCA2 mutations (12). Criteria for Genetic Risk AssessmentPatients with greater than an approximate 20-25% chance of having an inherited predisposition to breast cancer and ovarian cancer and for whom genetic risk assessment is recommended (13):
*Cancer of peritoneum and fallopian tubes should be considered a part of the spectrum of the HBOC syndrome. Close relative is defined as a first-degree relative (mother, sister, daughter) or second-degree relative (grandmother, granddaughter, aunt, niece). Patients with greater than an approximate 5-10% chance of having an inherited predisposition to breast cancer and ovarian cancer and for whom genetic risk assessment may be helpful (13):
HBOC Syndrome Risk Scoring ToolWe have developed a simple numerical teaching tool that can be used to estimate a patient's candidacy for BRCA testing. This tool approximates NCCN guidelines by assigning 1 or 2 points to each person with each "red flag" relevant cancer in the three-generation family tree. The patient's points are added together with the maternal points and then again with the paternal points. A sum of 0 point would indicate a sporadic (low) classification. A sum of 1 point would usually indicate a familial (medium) risk classification; however, a 1-point patient may still qualify for genetic testing if there is a limited family structure or an ethnic predisposition to BRCA mutations. A sum of >2 points will generally qualify for BRCA testing, although combinations that involve third-degree relatives may be evaluated to determine if testing is warranted. This tool should only be used as an estimate, and not a conclusive testing guide. Circle each cancer point that applies, then total a+b and a+c
Each primary cancer counts separately
HBOC, hereditary breast and ovarian cancer; LFS, Li-Fraumeni syndrome 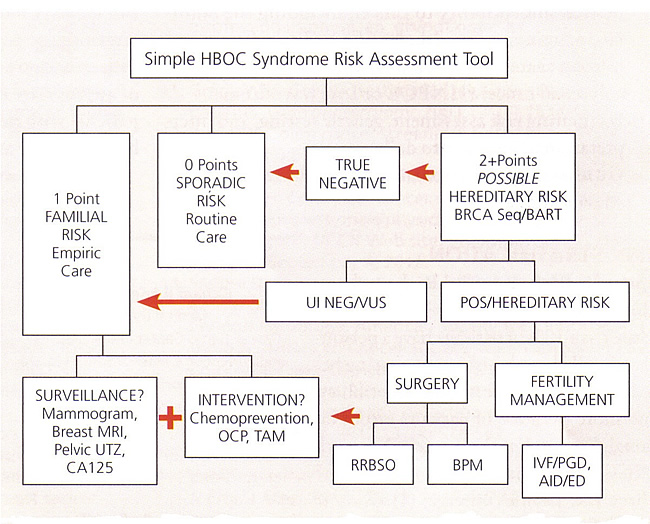 Abbreviations: AID, artificial insemination by donor; BART, BRCA Analysis rearrangement test; BPM, bilateral prophylactic mastectomy; CA 125, cancer antigen 125; ED, egg donation; HBOC, hereditary breast and ovarian cancer; IVF, in-vitro fertilization; MRI, magnetic resonance imaging; OCP, oral contraceptive pills; PGD, preimplantation genetic diagnosis; POS, positive; RRBSO, risk-reducing bilateral salpingo-oophorectomy; TAM, tamoxifen; UI NEG, uninformative negative; UTZ, ultrasound; VUS, variant of uncertain significance. Genetic Counseling to Reduce Risk of Breast CancerGenetic counseling should include a discussion of possible outcomes of testing specifically addressing the issues of positive, negative, and uninformative test results, or variants of unknown significance. Options for surveillance, chemoprevention, and risk-reducing surgery should be discussed before testing. Possible psychologic and familial implications of test results should also be considered. Written materials may help individuals share information with relatives about their potential genetic risks. The genetic counseling session should also include a discussion of the cost of genetic testing. Many insurance companies, including Medicare, will cover a significant portion of the expense for certain individuals. Medicare and other insurance companies have written guidelines for covering the cost of genetic testing. An important aspect of genetic counseling is discussion of current legislation regarding genetic discrimination and the privacy of genetic information. The federal Genetic Information Nondiscrimination Act of 2008 protects individuals against health and employment discrimination based on genetic information. Many states also have state laws that provide similar protection. These laws do not apply to other forms of insurance, which may include life or disability insurance. Ideally, it makes sense to initiate testing in an affected individual. If, however, no affected individual is available, genetic testing may still provide useful information. If a deleterious mutation is identified, patients at risk can be appropriately counseled about screening or other risk-reduction approaches. If no deleterious mutation is identified, patients need to be counseled that this could be because of one of several possibilities including (a) a deleterious mutation present in family that the patient did not inherit; (b) an undetectable mutation in BRCA1 and BRCA2, or a yet unidentified cancer susceptibility gene is present in the family, and it remains unknown whether the patient share this predisposition; or (c) no inherited predisposition in the family. Current strategies to reduce the risk of developing ovarian cancer or fallopian tube cancer in women at high risk with known deleterious BRCA mutations include surveillance, chemoprevention, and surgery. Given the extremely high risk for ovarian cancer and fallopian tube cancer in women with mutations in BRCA1 and BRCA2, consensus groups have recommended periodic screening with CA 125 and transvaginal ultrasonography, beginning between the ages of 30 years and 35 years or 5-10 years earlier than the earliest age of first diagnosis of ovarian cancer in the family (14). The low prevalence of ovarian cancer and the high likelihood of a positive screening test result necessitating further invasive surgical evaluation are obstacles in ovarian cancer screening programs among women in inherited risk (15). It is hoped that future ovarian cancer trial will benefit from better serum markers and improved screening algorithms to enhance the ability to discriminate between normal and abnormal CA 125 values and ultrasound findings. The benefits and magnitude of reduced risk with oral contraceptives for women with a BRCA mutation has not been reported as consistently as for the general, low-risk population. It is reasonable for women with mutations in BRCA1 and BRCA2, to use oral contraceptive. Most studies report a reduced risk of ovarian cancer among those who used oral contraceptives for a longer duration (more than 3 to 6 years) (16). The relative risks and benefits for both chemoprevention and reproductive control should be carefully weighed by the patient and her physician. Prophylactic Bilateral Salpingo-oophorectomy (BSO)It is strongly recommended in women who carry BRCA mutations because of the high mortality rate of ovarian cancer and the lack of effective screening and prevention approaches (17). Fortunately, the risk of hereditary breast and ovarian cancer does not rise dramatically until the late 30s in women with BRCA1 mutations, and the late 50s for women with BRCA2 mutations (18), so women have the opportunity to complete their families before undergoing BSO. The past practice of performing prophylactic surgery based solely on family history should largely be abandoned. Half of women in families with BRCA mutations would not be expected to be carriers. Currently, the decision to proceed with prophylactic BSO surgery is based primarily on the results of BRCA mutational analysis. There is some evidence that certain types of BRCA mutations may predispose more strongly to ovarian cancer, but this observation has not been consistently confirmed (19). Although BSO is a major surgical procedure, studies show that most high-risk women undergoing genetic testing accept prophylactic bilateral salpingo-oophorectomy. Laparoscopy and laparotomy are both options for risk-reducing BSO. For both procedures, a thorough inspection of peritoneal surfaces is necessary. Peritoneal washings should be obtained. The diaphragm, liver, omentum, bowel, paracolic gutters, and appendix are inspected in the abdomen. The ovaries, fallopian tubes, uterus, bladder serosa, and cul-de-sac are inspected in pelvis. Any abnormal areas should undergo biopsy. The ovarian vessels should be isolated and ligated proximal to the end of identifiable ovarian tissue to ensure that all ovarian tissue is completely removed. If a hysterectomy is not being performed, the fallopian tube should be divided at its insertion into the uterine cornu. When performing a laparoscopic procedure, to optimize preservation of the ovarian surface epithelium, the specimens can be placed in an endoscopic bag before removal from abdomen. The decision to perform a concurrent hysterectomy should be individualized. Arguments in favor of hysterectomy include a more simplified hormone therapy strategy (with estrogen only) and a theoretical increased risk of cancer in the corneal fallopian tube (20). In addition, hysterectomy may be considered when there are other medical indications for removal of the uterus and cervix. For women taking tamoxifen, hysterectomy may be considered to reduce their endometrial cancer risk (21). Pros and Cons of Risk-reducing Salpingo-oophorectomy
Cons:
What Age to Consider Risk-reducing Salpingo-oophorectomy?Risk-reducing and elective salpingo-oophorectomies are removal of the ovaries and fallopian tubes for the potential benefit of preventing long-term morbidity and mortality. The term risk-reducing salpingo-oophorectomy implies that the ovaries are normal at the time of removal. Women with BRCA1 or BRCA2 mutations should be offered risk-reducing salpingo-oophorectomy by age 40 years, or when childbearing is complete (13). Ovarian cancer will be diagnosed in less than 2-3% of women with BRCA1 or BRCA2 mutations before age 40 years. For women with BRCA1 mutations, the risk of ovarian cancer markedly increases during the 40s, with 10-21% of BRCA1 mutation carriers developing ovarian cancer by age 50 years. The risk of premenopausal ovarian cancer is much lower in BRCA2 mutation carriers, with no more than 3% of BRCA2 mutation carriers developing ovarian cancer by age 50 years (13),(22). Given the different timing of ovarian cancer risk, consideration can be made for counseling patients with BRCA1 mutations differently than for BRCA2 mutation carriers. However, women with BRCA2 mutations have a 26-34% chance of developing breast cancer by age 50 years (18),(22), and the maximum benefit of removing the ovaries on breast cancer risk reduction is achieved the earlier the ovaries are removed (23). Given these issues, the timing of risk-reducing salpingo-oophorectomy should be based on individual patient needs, taking into consideration their desire to preserve fertility or prevent premature surgical menopause with the age-dependent impact of risk-reducing salpingo-oophorectomy on both breast cancer and gynecologic cancer risks. Prophylactic Salpingectomy and Delayed OophorectomyProphylactic bilateral salpingo-oophorectomy is advised for women with BRCA mutations, but there are adverse consequences of premature menopause. The majority of BRCA-associated ovarian cancers appear to arise in the fallopian tube; therefore, salpingectomy may be an alternative to bilateral salpingo-oophorectomy. With the understanding that perhaps 60% of pelvic serous cancers may originate in the fallopian tube, there has been a more recent discussion of risk-reducing salpingectomy as a potential alternative to risk-reducing BSO for the treatment of BRCA mutation carriers, particularly in women reluctant to undergo risk-reducing BSO (39). The authors publish their Markov Monte Carlo simulation comparing risk-reduction BSO at 40 years of age with both risk-reduction salpingectomy at 40 years of age followed by delayed bilateral oophorectomy at 50 years of age (40). This thoughtful model predicts the expected reduction in pelvic serous cancers, breast cancers, and incremental deaths due to cardiovascular disease. The conclusion the study was bilateral salpingo-oophorectomy often the greatest risk reduction for breast and ovarian cancer among BRCA mutation carriers. However, when considering quality-adjusted life expectancy, bilateral salpingectomy with delayed oophorectomy is a cost-effective strategy and may be an acceptable alternative for those unwilling to undergo bilateral salpingo-oophorectomy (40). It is important to emphasize that the standard of care for women inheriting germline mutations in BRCA1 and BRCA2 still remains prophylactic BSO after completion of childbearing or around the age of 40 years. It offers the greatest risk reduction in breast and ovarian cancer compared with salpingectomy with or without delayed oophorectomy. However, a significant proportion of women do not undergo BSO, and many choose surveillance alone for ovarian cancer despite the limited benefit of existing screening methods (41). Ovarian cancer drives the mortality rate among BRCA mutation carriers, the therefore any intervention that reduces ovarian cancer risk is likely to be validated prospectively, bilateral salpingectomy with delayed oophorectomy may be a reasonable alternative to BSO, especially for those who are reluctant to undergo the latter procedure because of the potential effect on quality of life. Management of Women with Strong Family History and Negative BRCA1 or BRCA2 MutationAlthough, in most cases, an inherited predisposition to ovarian cancer is caused by mutations in BRCA1 or BRCA2, current technology does not allow identification of all mutations that must exist in these genes (24). Additionally, linkage studies have suggested that is less than one half of families with four or more cases of breast cancer, but no cases of ovarian cancer (families with site-specific breast cancer), breast cancer are caused by BRCA1 or BRCA2 mutation (25). Given these issues, women with a personal or family history of breast cancer who have tested negative for a BRCA mutation should be managed based on their family history. Preliminary data have suggested that women from families with a history of site-specific breast cancer in which no BRCA mutation is identified remain at a significantly increased risk of breast cancer, but may not be at a significantly increased risk of ovarian cancer (26). It is important for high-risk individuals to stay in contact with clinicians experienced in the care of women at increased risk, given rapidly developing research and refinements in testing technology. For example, a test for large rearrangements in the BRCA1 and BRCA2 genes has been developed that may help to identify mutations in a small percentage of the high-risk families who previously tested negative for these genes. Reproductive OptionsThe outcomes of genetic testing can have profound impact on family planning decisions for individuals of reproductive age who are found to be carriers of BRCA1/2 mutations. For example, in cases where both partners carry a BRCA2 mutation, there may be a high risk for the offspring to develop a rare Fanconi anemia/brain tumor phenotype (recessive disorder). Counseling for reproductive options such as prenatal diagnosis, preimplantation genetic diagnosis (PGD) and assisted reproduction may therefore be warranted for couples expressing concern over the BRCA mutation carrier status of their future offspring. Such counseling should include a comprehensive discussion of the potential risks, benefits, and limitations of reproductive options. Prenatal diagnosis involves post-implantation genetic analysis of an early embryo, utilizing chorionic villi or amniotic fluid cell samples; genetic testing is typically conducted between week 12 and week 16 of gestation, and testing results may potentially lead to a couples decision to terminate pregnancy (27). During the past two decades, PGD has emerged as an alternative method of genetic testing in early embryos. PGD involves the testing of 1 or 2 cells from embryos in very early stages of development (i.e., 6 to 8 cells) after in vitro fertilization (IVF). This procedure allows for the selection of unaffected embryos to be transferred to the uterus, and may, therefore, offer the advantage of avoiding potential termination of pregnancy. However, procedures such as PGD are not without limitations as it may still require a confirmatory prenatal diagnosis depending upon a couples medical needs or requests. Moreover, the PGD process requires the use of IVF regardless of the fertility status of the couple (i.e., also applies to couples without infertility issues), and IVF may not always lead to a successful pregnancy. Lastly, the technology or expertise may not be readily available in a couples geographical location. Various factors, both medical and personal, must be weighed in the decision to utilize prenatal diagnosis or PGD. Medical considerations may include factors such as the age of onset of the hereditary cancer, penetrance, severity or associated morbidity and mortality of the cancer, and availability of effective cancer risk reduction methods or effective treatment. Although the use of prenatal diagnosis or PGD is relatively well established for severe hereditary disorders with very high penetrance, their use in conditions associated with lower penetrance (e.g., hereditary breast or ovarian cancer syndrome) remains somewhat controversial from both an ethical and regulatory standpoint. Personal considerations for the decision to utilize prenatal diagnosis or PGD may include individual ethical beliefs, value systems, cultural and religious beliefs, as well as social and economic factors. Based on results from surveys administered to women at high risk for hereditary breast or ovarian cancer, 50%-75% of respondents felt that PGD was an acceptable option for high-risk individual yet only about 14%-33% would consider undergoing PGD themselves (28). Importantly, the surveys suggested that the majority of high-risk women have little or no knowledge of PGD, highlighting the need for better awareness and education regarding potential reproductive options. Successful births have been reported with the use of PGD and IVF in BRCA1/2 mutation carriers, but data in the published literature are still very limited. In addition, data pertaining to long-term safety or outcomes of PDG and assisted reproduction in BRCA mutation carriers are not yet available. Hereditary Nonpolyposis Colon Cancer (HNPCC) or Lynch SyndromeIt is an autosomal dominant inherited disease of the DNA mismatch repair system. Many authorities refer to HNPCC as the Lynch syndrome, believing that HNPCC fails to adequately acknowledge the importance of extracolonic malignancies, such as endometrial, ovarian, upper gastrointestinal tract, and urinary tract cancers associated with this disease. This syndrome accounts for 5-10% of all colon and endometrial cancers (29). Linkage analysis of high-risk families led to the discovery that Lynch syndrome is caused by germ-line mutations in a class of genes responsible for repairing certain types of DNA mutations (29). These genes are called mismatch repair genes and give rise to proteins that proofread DNA and correct mistakes that are made during the normal process of replication. MSH2 (MutS homolog 2) and MLH1 (MutL L homolog 1) are the most commonly mutated mismatch repair genes in this syndrome. MSH2 and MLH1 are located on chromosomes 2p16 and 3p21, respectively. Germ-line mutations in other mismatch repair genes (MSH6, PMS1, and PMS2) have been identified, but at a lower frequency (29). Likelihood of Developing a Cancer if there is Germ-line MLH1 or MSH2 Mutation:
Family history is the first step in determining whether a patient is at increased risk for an inherited disease. Several relatively stringent criteria have been developed to identify individuals at high risk for Lynch syndrome. About 30% of families fulfilling the Bethesda criteria and 50-92% of families fulfilling the Amsterdam criteria will have germ-line DNA mismatch repair gene mutation (30),(31). The Bethesda criteria for HNPCC (see below) seem to be the most sensitive in predicting mismatch repair mutations in HNPCC families (30). Any of the following:
The Amsterdam II criteria (see below) for HNPCC are more specific (31). All of the following:
HNPCC / Lynch Syndrome Simple Risk Assessment Tool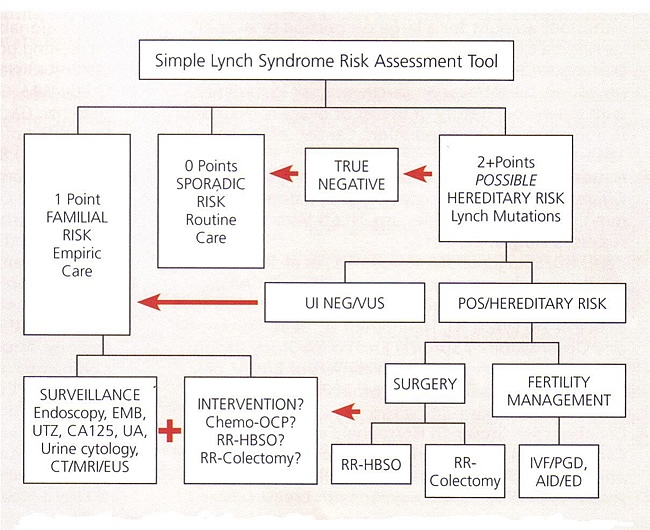 Abbreviations: AID, artificial insemination by donor; CA 125, cancer antigen 125; CT, computed tomography, ED, egg donation; EMB, endometrial biopsy; EUS, endoscopic ultrasound; HBSO, hysterectomy with bilateral salpingo-oophorectomy; IVF, in-vitro fertilization; MRI, magnetic resonance imaging; OCP, oral contraceptive pills; PGD, preimplantation genetic diagnosis; POS, positive; RR, risk-reducing; UA, urinalysis; UI NEG, uninformative negative; UTZ, ultrasound; VUS, variant of uncertain significance. Surveillance for Hereditary Nonpolyposis Colon Cancers (HNPCC)When the mutations are identified, a simpler, less costly, genetic analysis can be performed on other family members. Extensive counseling is recommended before and after the testing. Once the genetic process has been completed, cancer risk can be assigned for the patient and her family. When a mutation has been identified, genetic testing should be offered to all family members. These individuals should each undergo genetic counseling so that the advantages and disadvantages of testing can be explained. Carriers of a mutation are offered intensive surveillance. If no mutation is identified in the patient, but the family meets Amsterdam criteria the protocol for Lynch syndrome, surveillance should observed. Mortality from colorectal cancers in this group has been shown to be reduced by colonoscopy screening (32). Most authorities believe that risk-reducing hysterectomy with bilateral salpingo-oophorectomy has a role in the management of these patients. Colorectal cancers in HNPCC syndrome have a propensity to occur in the proximal colon, relative to sporadic cases, which are more often distal. Prophylactic colectomy is recommended to patients with HNPCC mutation in whom polyps are identified at a young age and also to patients who have polyps that are microsatellite instability-positive or those individuals who are unable to undergo regular surveillance. Chemoprevention using non-steroidal anti-inflammatory agents in this group of patients to prevent colon polyps is currently under study, and data should be forthcoming. If these patients are being explored for another reason, consideration should be given to counseling regarding possible risks and benefits of a total hysterectomy with bilateral salpingo-oophorectomy. Surveillance for HNPCC (30),(33):
GI, gastrointestinal Li-Fraumeni SyndromeLi-Fraumeni syndrome (LFS) is associated with germline mutations in the tumor suppressor gene TP53, which cause a highly penetrant predisposition to develop sarcomas as well as early-onset breast, brain, adrenocortical, and other cancers. People with LFS have an absolute risk for developing any cancer of approximately 50% by age 30 years and 90% by age 60 years (34). LFS account for up to 1% of breast cancer in United States; such cancers may have a distinctive phenotype, expressing hormone receptors and HER2/neu. The de novo mutation frequency is estimated at 7% to 20%, such that TP53 testing may be appropriate for selected patients in the absence of a family cancer history (34). Cowden SyndromeCowden syndrome (CS) conveys increased susceptibility to breast, thyroid, endometrial, and colon cancers, and benign hamartomas affecting multiple organs. It is an autosomal dominant condition associated with mutations in the tumor suppresser gene PTEN. Gynecologic malignancies are common with a 5-10% risk of endometrial cancer and 25-50% risk of breast cancer (42). Outward manifestations include macrocephaly and papillomatous papules on the face, oral-lingual mucosa, and extremities, which are almost ubiquitous by age 30 years. The lifetime breast cancer risk is 25-50%, with an average age of diagnosis of 38-46 years (35). The life-time risk for endometrial cancer is 5-10% (35). Germline mutation in the PTEN gene are identified in approximately 80% of the patients meeting clinical diagnostic criteria, almost half of whom have no family history of CS. The reported frequency of CS is 1 in 200,000, but it is likely under-diagnosed (35). Recurrent, multiple endometrial polyps portend a high risk of endometrial cancer in women with CS. Monitoring for malignancy and consideration of hysterectomy after childbearing is completed is warranted (42). Hereditary Diffuse Gastric Cancer SyndromeHereditary diffuse gastric cancer syndrome is associated with mutations in the CDH1 gene and is characterized by an 80% lifetime risk of developing gastric cancer (36). Women with CDH1 mutations have up to a 60% lifetime risk for developing lobular breast cancer; however, the prevalence of CDH1 mutations in women selected for early age at onset or family history of lobular breast cancer is low, about 1% (36). Among families with diffuse gastric and lobular breast cancer, CDH1 mutations are identified in 25-50% (36). Peutz-Jeghers SyndromePeutz-Jeghers syndrome (PJS), resulting from alterations in the STK11 gene, features hamartomatous polyps throughout the gastrointestinal tract, leading to intussusception and bowel obstruction. STK111 mutations convey increased risks for breast, ovarian, cervical, pancreatic, gastric, and colon cancer, in addition to benign sex cord tumors. Breast cancer risk reaches 8% by age 40 years and 32% by age 60 years. Half of STK11 mutation carriers lack a family history of PJS (37). Neurofibromatosis-1Neurofibromatosis-1 (NF-1) features cutaneous neurofibromas and malignant peripheral nerve sheath tumors, learning disabilities, and a prevalence of 1 in 3,000 live births (38). A recent study of women with NF-1 reported five-fold increased risk for developing breast cancer before age 50 years and a 3- to 4-fold increase in lifetime cancer risk (38). Advancing Standard of Cancer CareAs screening for hereditary cancers has become more readily available, many questions surrounding liability, risk management, and patient safety have emerged. As in all medico-legal issues, these areas of concern generally pertain to standard of care, documentation, consent, patient expectations, and follow up. Many providers feel that hereditary cancer screening is not standard of care in the primary care office. However, there are three points that are very important to remember. It is standard of care to obtain a comprehensive and complete family history and update it on a routine basis. It is standard of care to give patients appropriate information based on that family history so that they can make educated decisions about their medical care. Finally, it is standard of care to thoroughly and completely record whatever was discussed with the patient. If you adhere to these three points, then it would seem that screening for hereditary cancer is, in fact, standard of care. Documentation: once you have identified someone that fits criteria for genetic testing, how much documentation is needed? Is it adequate to have your note state: "information on genetic testing given" or "brochure given?" although it is nice to see your plan documented, it is much more important to see the reasoning behind the plan. In this instance, an expanded note such as: "based on family history, genetic testing recommended. Patient understands that if the test is positive there is a substantial increase in the risk of ovarian and/or breast cancer or [the particular Lynch syndrome cancer you are screening for]." Although we know that we discussed cancer risks, the patient can easily contradict what is not documented in their chart. Patients may argue that if they understood their risks, they would, of course, have consented to the test. Incorporating some sort of tracking system into your office is prudent. This can allow for you to follow-up with a patient after she has been referred for genetic counseling. Without this type of tracking and follow up, a troubling question can be raised: "if you felt it was important enough for the patient to have this testing, why was not it important enough for you to see if the test was done?" Informed Consent: Informed consent or informed refusal need to be addressed when discussing hereditary risk assessments. Typically, informed consent has dealt with only with informing patients of risks associated with invasive procedures. However, there has been an expansion of what adequate informed consent includes. As part of adequate informed consent we are now asked to give all treatment options, along with the risks and benefits of each option. Therefore, if we do not give appropriate patients the option of genetic testing (along with its risks and benefits), we may be found to be negligent on a consent basis should there be an adverse event. This is where informed refusal may come into play, if a patient does not want to do what the provider feels is appropriate, or has not followed up with a genetic counseling referral when you referred her to one, documenting their refusal, or lack of follow-up, may ultimately be more important than documenting their consent. Informed refusal documents that the physician has done what is prudent and that it is the patients choice to not follow through. Many states have some element of contributory negligence, and this can go a step further and document the reason for the patients refusal; fear of the test result, unwillingness to do anything about the result, or financial reasons may be part of a patients decision to refuse testing. Presently, one of the major causes of malpractice cases involves issues with breast cancer. Typically, allegations include both delayed diagnosis and failure to diagnose. We are now seeing a new allegation that is being referred to as a failure of our "duty to inform" or "duty to warn". This pertains to the failure to identify a patient at risk for a hereditary cancer so that increased surveillance could have been implemented to diagnose the cancer earlier or that risk-reducing or prophylactic surgery could have been performed. These types of cases will be very difficult, if not impossible, to defend without proper documentation, including documentation of a patients refusal of testing, and documentation of the explanation of very specific cancer risks. SummaryMore than 300,000 US women are estimated to carry a high inherited risk for developing breast and ovarian cancer. An increasingly broad array of genetic tests is available to define patient's risk, yet results still may prove inconclusive or complicated to interpret. Cancer genetics professionals offer comprehensive risk assessment, counseling, and management recommendations for patients and families with an inherited predisposition for developing breast and ovarian cancer. As specialists in womens health, hereditary risk assessment is our responsibility. A focused cancer family history should be part of initial evaluation of all patients. Maternal and paternal pedigrees should be constructed to include at least three generations (the patient's, her parents', and her grandparents' generations). Information regarding the family member's age at diagnosis and additional details such as breast cancer in both breasts are very helpful. Updates of the family history should be asked about at annual exams because additions and new diagnoses may alter the initial recommendation regarding genetic testing. A genetic risk assessment is recommended for patients with a greater than an approximate 20-25% chance of having an inherited predisposition to breast cancer and ovarian cancer. Women with BRCA1 or BRCA2 mutations should be offered risk reducing salpingo-oophorectomy by age 40 years or when child-bearing is complete. For a risk-reducing bilateral salpingo-oophorectomy, all tissue from the ovaries and fallopian tubes should be removed. Thorough visualization of the peritoneal surfaces with pelvic washings should be performed. Complete, serial sectioning of the ovaries and fallopian tubes is necessary, with microscopic examination for occult cancer. References
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||