


Principes de conseil génétique et diagnostic prénatal
Dr Francis H. Boudreau
Président, Département d'obstétrique et de gynécologie
St. Elizabeth's Medical Center, Boston, MA (USA)
En collaboration avec Women's Health & Education Center (WHEC)
La disponibilité d'un diagnostic prénatal pour une vaste gamme de troubles a été une avancée majeure dans le domaine de la génétique de la reproduction. L'échographie a joué un rôle central dans le développement de diverses approches de diagnostic prénatal. En outre, l'intégration d'un programme axé sur la génétique de diagnostic prénatal par échographie tertiaire a été montré pour augmenter la précision du diagnostic par rapport à l'échographie seule.
Le but de ce document est de mettre l'accent sur l'épidémiologie des anomalies génétiques et inclure une description des différentes procédures de diagnostic prénatal en cours d'utilisation.
Épidémiologie
Toutes les grossesses entraînent un risque de base des malformations congénitales. La plupart des généticiens cite un chiffre de 3% à 4% pour les malformations congénitales majeures. Il est important de garder ce chiffre de référence à l'esprit lorsque les couples counseling au sujet de leur risque d'un défaut spécifique. Les étiologies des défauts de naissance peut être décomposé en 4 catégories:
- Chromosomiques
- Seul gène ou mendélienne troubles
- Multifactorielle des troubles
- Tératogènes ou des effets environnementaux
Les malformations sont les défauts de la structure d'un organe ou région du corps résultant d'un processus intrinsèquement anormale de développement. Ces malformations mai être uniques ou multiples, elles comprennent, par exemple, des anomalies congénitales causées par des anomalies monogéniques. Déformations sont les anomalies dans la forme, la forme, ou la position causés par des forces mécaniques. De telles déformations surviennent souvent avant la naissance à la suite de moulage intra-utérin ou de contrainte. Les facteurs conduisant à des déformations mai être extrinsèque ou intrinsèque. Perturbation est une anomalie morphologique résultant de la dégradation des tissus préalablement normale. Les perturbations peuvent être causées par des forces extrinsèques, l'ingérence interne avec un processus de développement ou d'insultes vasculaire. Exemples de perturbations incluent amputations dues à des bandes amniotique, et gastroschisis et porencéphalie, à la fois pensé qu'il résulte de la insultes utero vasculaires. La séquence est un modèle d'anomalies multiples résultant d'une anomalie primaire unique ou un facteur mécanique, elle mai être une malformation, déformation ou perturbations. Un exemple est la séquence Potter, dans lequel oligohydroamnios de n'importe quelle cause conduit à des fonctions similaires de compression du foetus: des traits faciaux caractéristiques, les malformations des mains et des pieds, et une hypoplasie pulmonaire. Syndrome est un modèle d'anomalies multiples connus pour avoir une communs spécifiques, étiologie. Un exemple est le syndrome de Down, qui est causée par la présence d'une copie supplémentaire du chromosome 21. Association est utilisé pour identifier l'apparition de deux ou plus de fonctionnalités le plus souvent que prévu par le seul hasard, mais pour lesquelles aucune étiologie a été démontrée. Un exemple courant est le VATER (vertébrale, Anal, trachéo-oesophagienne, et des anomalies rénales) association. Ce terme n'implique pas un diagnostic précis, mais invite plutôt de la recherche pour des défauts particuliers quand un autre volet de l'association est identifiée.
I. anomalies chromosomiques:
Anomalies chromosomiques surviennent chez environ 0,5% des nouveau-nés et il est bien connu que le risque pour de nombreuses anomalies chromosomiques augmente avec l'âge maternel (1). Les études sur l'origine prénatale du chromosome 21 dans le syndrome de Down ont montré que 95% des cas de trisomie 21 sont dus à la non-disjonction maternelle et seulement environ 5% des cas contiennent un supplément de 21 paternel. Bien que l'âge maternel est fortement corrélée avec le risque du syndrome de Down, la plupart des enfants atteints du syndrome de Down sont nés de femmes de moins de 35 ans. Beaucoup d'autres anomalies chromosomiques sont également associés avec l'augmentation de l'âge maternel, y compris 13 et 18, 47, XXX, 47, XXY et 47, XYY. Structural anomalies chromosomiques, telles que les translocations et délétions, ainsi que le syndrome de Turner (45, X) ne semblent pas être liés à l'âge.
Les anomalies chromosomiques sont la principale cause connue de la perte de la grossesse, et au moins 95% des chromosomes anormaux conceptions sont perdus avant le terme. On estime qu'un minimum de 10% à 15% des conceptions sont des anomalies chromosomiques. Études de fausses couches montrent qu'environ 50% de ces anomalies chromosomiques sont des trisomies, 20% sont monosomies, 15% sont triploïdes, et le reste est tétraploïdes et des anomalies structurelles. Certaines anomalies chromosomiques, telles que la trisomie 16, sont communes à la conception, mais jamais vu en live-nés.
Anomalies chromosomiques de l'nés vivants (2):
| Type d'anomalie | Incidence |
|---|---|
| Numerical aberrations: les chromosomes sexuels 47, XYY | 1 / 1080 naissances masculines |
| Autosomal trisomies 13 | 1 / 19, 000 |
| Structurel équilibré anomalies: Robertsonienne t (Dq; Dq) | 1 / 1500 |
| Déséquilibrée des anomalies structurelles: Robertsonienne | 1 / 14, 000 |
II. Seul gène Disorders:
Seul gène, ou mendélienne, les troubles se produisent dans environ 1% des nouveau-nés. Un grand nombre de troubles mendélienne ont été décrites, bien que l'incidence d'un trouble spécifique en général est rare. Par exemple, la fibrose kystique a une fréquence porteuse d'environ 1 sur 20, entraînant une fréquence de la maladie d'environ 1 en 1600. Parmi les caractères génétiques reconnus unique, plus de la moitié sont à transmission autosomique dominante, un tiers environ sont autosomique récessive, et le reste est lié à l'X.
Troubles autosomique récessif: survient plus fréquemment chez les personnes dont les parents sont tous deux porteurs sains du gène récessif même. Le risque pour la descendance de ces parents est de 25% à chaque grossesse, et ces troubles surviennent le plus souvent sans antécédents familiaux. Autosomique récessive troubles sont souvent graves, et beaucoup sont des troubles métaboliques. Les transporteurs devront en moyenne 50% de l'activité enzymatique. Le risque accru de parents qui sont cousins germains d'avoir un enfant souffrant de graves malformations est d'environ 6%, soit deux fois le risque de fond.
Troubles autosomique dominante: les personnes touchées sont hétérozygotes pour un allèle anormal, qu'ils transmettent à 50% de leur progéniture, mâle ou femelle. La population non infectée ne transmet pas le désordre. Âge d'apparition d'un grand nombre de ces troubles est variable et les personnes ayant un gène défectueux, qui sont donc destinés à développer la maladie, mai être asymptomatique jusqu'à l'âge adulte. Les personnes à risque mai ont leurs propres enfants avant l'âge auquel ils développent des symptômes. La sévérité de nombreux troubles autosomique varie considérablement, même au sein d'une famille. La gravité, susceptible de descendance atteinte est difficile à prédire, et un parent légèrement touchés mai ont un enfant gravement touchés. Nouvelles mutations pour les maladies autosomiques dominantes semblent être plus fréquents avec l'âge paternel. En raison du grand nombre de conditions autosomique dominante qui peut résulter de telles mutations, aucun test prénatal spécifique n'est disponible. Dans la neurofibromatose environ 50% des cas sont dus à de nouvelles mutations. En achondroplasie, la forme homozygote est létal et peut être prévu de se produire dans 25% des cas les deux parents sont touchés. De descendants tels, 50% seront hétérozygotes et touchés, et 25% sera normal.
X-troubles liés: la plupart des troubles liés à l'X sont récessives, avec hémizygotes (ayant une seule copie du chromosome X) hommes affectés et hétérozygotes femmes porteuses affectées. Une femme porteuse va transmettre le gène mutant à 50% de sa progéniture, donc, la moitié de ses fils seront touchés et la moitié de ses filles seront porteuses. Un homme atteint passera le chromosome X muté à l'ensemble de ses filles (qui sont donc obliger les transporteurs) et à aucun de ses fils (qui n'héritent que son chromosome Y). Dystrophie musculaire de Duchenne mai être graves ou mortels au cours de la petite enfance, afin que les personnes touchées ne se reproduisent pas. X-troubles liés peuvent également être dominant, bien que ce type de transmission est beaucoup moins fréquente.
III. Hérédité multifactorielle:
Malformations congénitales isolées, comme les pieds bots, une fente labiale et des malformations du tube neural (ATN) sont parmi les anomalies génétiques les plus courantes et sont hérités par une combinaison de facteurs génétiques et environnementaux. Exemples de caractères multifactoriels sont: hydrocephly, anomalies du tube neural, fente labiale avec ou sans fente palatine, fente palatine, anomalies cardiaques, hernie diaphragmatique, une omphalocèle, une agénésie rénale, les défauts de fusion de Müller, les défauts de réduction des membres, la sténose du pylore et pied bot varus équin.
Le risque de récurrence pour la famille d'un membre de la famille touchée est généralement plus élevé que le risque pour la population, la plupart des affections multifactorielles ayant un risque de récurrence de 2% à 5% chez les parents au premier degré. Ce risque est accru si plusieurs membres de la famille sont touchés. Il existe également un risque accru de troubles multifactorielle avec consanguinité parentale. Caractères multifactoriels tombent généralement dans l'une des trois catégories suivantes: seuil variable en continu ou des troubles complexes. Traits continus ont tendance à être moins extrêmes dans la descendance des personnes touchées, en raison du principe statistique de la régression à la moyenne. Ce sont généralement mesurables ou quantitative traits comme la hauteur ou taille de la tête, et on pense qu'ils résultent des effets individuellement petites de nombreux gènes combinés avec des facteurs environnementaux. Traits de seuil, ne semble pas jusqu'à un certain seuil de responsabilité est dépassé. Fente labio-palatine et une sténose du pylore sont des exemples de traits de seuil. Traits certain seuil ont une prédilection pour un sexe, ce qui indique que les mâles et les femelles ont un seuil de responsabilité différents. Le risque de récurrence pour les traits de seuil est également plus élevé si le défaut est grave, ce qui indique aussi la présence de plus de gènes anormaux ou influences. Par exemple, le risque de récurrence après la naissance d'un enfant avec une fente labio-palatine bilatérale est de 8%, contre seulement 4% après une fente labiale unilatérale, sans fente palatine. Troubles complexes de la vie adulte sont des traits dans lequel plusieurs gènes de susceptibilité d'un individu à des facteurs environnementaux, avec une maladie résultant de la combinaison la plus défavorable des deux. Cette catégorie comprend les maladies courantes, telles que les maladies cardiaques et l'hypertension (3).
IV. Tératogènes ou de l'exposition environnementale:
Tératogènes ou les expositions environnementales sont soupçonnés de provoquer des anomalies congénitales chez moins de 1% des nourrissons. Cette zone est importante parce que ces expositions sont potentiellement évitables. Il s'agit notamment des médicaments tératogènes, l'exposition à des agents tels que les rayonnements ionisants, et sous-produits de certains troubles maternels métaboliques comme le diabète et la phénylcétonurie.
Pour plus de détails s'il vous plaît examiner le chapitre sur: les drogues pendant la grossesse et l'allaitement
Indications pour le diagnostic prénatal:
Il est essentiel que les couples à risque être informé de toutes leurs options en matière de reproduction. Avant de tenter le diagnostic prénatal d'une maladie héréditaire, il est essentiel d'établir ou de confirmer la direction en vue d'éliminer la possibilité d'un diagnostic erroné dû à phénotypique, métabolique ou génétique hétérogénéité. Dans le cas d'un diagnostic enzymatique ou moléculaire, le défaut précise doit être démontrée dans le probant ou des parents concernés. Si le proposant est décédé, l'hétérozygotie des deux parents (ou de la mère pour une maladie liée à l'X) doivent être documentées. Dans le cas d'un syndrome dysmorphique, toutes les manifestations de la maladie doivent être connus et cherché à l'époque de l'évaluation.
Les indications les plus fréquentes pour les études de diagnostic prénatal sont: l'âge maternel avancé, le dépistage biochimiques anormaux; enfant présentant une maladie chromosomique; porteur de la translocation équilibrée pour une maladie chromosomique; enfant présentant un trouble métabolique héréditaire; couples hétérozygotes détectés de façon prospective par des programmes de dépistage; précédente enfant avec un trouble détectable par l'échographie.
Multifactorielle des troubles: enfant présentant une anomalie du tube neural; élévation sérique en alpha-foeto-maternelle protéine (AFP) et de l'enfant précédent avec défaut de développement ou de syndrome de malformation décelable à l'échographie.
Environmental défauts: l'exposition prénatale aux drogues tératogènes ou agent infectieux.
L'utilisation d'un âge maternel supérieur à 35 ans ou plus au moment de la livraison ou plus au moment de la livraison à titre indicatif, pour les tests invasifs (amniocentèse ou prélèvement de villosités choriales), environ 20% des cas de syndrome de Down seront détectées. 80% des cas de syndrome de Down se produira dans le non-testés groupe. Dans un effort pour augmenter le taux de détection du syndrome de Down, midtrimester Marqueurs sériques maternels biochimique a été développée qui permet de détecter environ 60% des cas de syndrome de Down dans la population générale (4).
Biochimiques de dépistage des malformations congénitales:
Certains des plus grands progrès de la génétique au cours des deux dernières décennies ont eu lieu dans le domaine du dépistage biochimique des malformations congénitales chez le ftus. Un certain nombre de marqueurs biochimiques ont été étudiés pour une utilisation dans le dépistage prénatal. Ce sont:
Alpha-fto-protéine (AFP):
L'AFP est une protéine onco-foetale produite par le foie foetal et le sac vitellin et est la protéine sérique majeur dans la vie foetale précoce. Il entre d'abord le liquide amniotique par diffusion à travers la peau immature du ftus et plus tard par l'urine du foetus. Les femmes non enceintes ont des niveaux de l'AFP qui sont à peine détectables (1 U / L), tandis que les niveaux normaux médiane de 16 à 18 semaines de gestation de 18 à 40 U / L. AFP sérique maternelle continue à augmenter jusqu'à 28 à 32 semaines de gestation. Chez le ftus présentant une anomalie comme l'anencéphalie ou spina-bifida, l'AFP entre dans le liquide amniotique en quantité accrue, conduisant à des niveaux plus élevés dans le sérum maternel ainsi. Les niveaux de l'AFP sont élevées dans le liquide amniotique et le sérum maternel uniquement lorsque de telles lésions sont «ouverts», c'est à dire lorsque le tissu neural est exposée ou est recouvert d'une fine membrane. Pratiquement 100% des lésions de l'anencéphalie sont ouvertes; environ 20% des cas de spina bifida sont fermées, de même qu'environ 82% des encephaloceles (5). Les délibérations de la sensibilité et la spécificité des tests de l'AFP sont généralement limités à des lésions ouvertes, comme il est supposé que les lésions fermées ne seront pas détectées.
Dans les laboratoires fournissant l'AFP le dépistage, les résultats sont exprimés en multiples de la médiane (MoM), qui permet l'expression de mesures dans une voie commune. La distribution de sérum maternel AFP (AFPSM) est log-gaussienne, avec une valeur médiane de 1,0 pour les grossesses normales, 3,8 pour les grossesses avec un spina-bifida ouvert, et 6,5 pour les grossesses affectée avec l'anencéphalie. Les deux seuils les plus couramment utilisés pour AFPSM sont de 2,0 et 2,5 MoM. À 2,0 MoM les plages de taux de faux positifs de 4% à 6%, et le taux de détection de spina bifida ouvert varie de 85% à 90%. Avec un seuil supérieur de 2,5 MoM les baisses de taux de faux positifs de 2% à 3% et le taux de détection diminue également de 75% à 80%. Femmes atteintes de pré-diabète gestationnel ont des valeurs AFPSM cette moyenne de 20% inférieur au deuxième trimestre. Lorsque le niveau AFPSM est au-dessus du seuil (soit 2.0 ou 2.5 MoM) l'exactitude de la datation est évaluée. Si datation est basée sur la dernière période menstruelle a été utilisé pour le calcul initial, et l'échographie doit être effectuée pour la datation précise. L'amniocentèse mai être indiquée pour identifier la cause de l'élévation à l'AFP.
Causes d'un AFPSM élevé: grossesse multiple; cession foetale; hémorragie fto-maternelle, des anomalies placentaires, des anomalies de l'utérus; maternelle tumeurs de l'ovaire ou hépatique. Fetal défauts structurels: les anomalies du tube neural - spina-bifida, l'anencéphalie, encephaloceles, ouvert anomalies de la paroi ventrale - omphalocèle, laparoschisis; néphrose congénitale; triploïdie; agénésie rénale bilatérale, troubles congénital de la peau - l'épidermolyse bulleuse, aplasie; polykystique autosomique récessive maladie rénale; tératome sacro-coccygienne; adénomatoide kystique du poumon.
Triple Marker Screening:
Un certain nombre d'autres marqueurs biochimiques ont été étudiés pour une utilisation dans le dépistage des aneuploïdies. Les deux estriol non conjugué (uE3) et la gonadotrophine chorionique humaine (hCG), ajouter la spécificité et la sensibilité à la deuxième projection trimestre biochimiques du syndrome de Down. Marqueur de dépistage Triple avec l'AFP, uE3, et l'hCG est maintenant offerte systématiquement pendant la grossesse. La plupart des centres utilisent un risque de 1 / 270 du syndrome de Down at midtrimester comme le seuil au-dessus duquel l'amniocentèse est offert, ce risque est équivalent à celui d'un 35-year-old woman. Comme pour le dépistage AFP, poids maternel, la race, le diabète, et le nombre de ftus présents sont tous considérés dans le calcul des risques.
Fetal échantillonnage des tissus et l'ADN Méthodologie:
Certaines maladies héréditaires ne sont exprimés que dans un tissu spécifique, et donc l'échantillonnage de ce tissu est nécessaire si le diagnostic prénatal est d'être faite. Foie ftal, la peau du ftus et les muscles du ftus ont été obtenus par biopsie guidée par ultrasons (6). Que des progrès sont accomplis dans l'élucidation de l'anomalie moléculaire spécifique dans un nombre toujours croissant de maladies génétiques, le diagnostic prénatal est possible par une analyse ADN. Comme l'ADN est présent dans toutes les cellules nucléées, les techniques minimalement invasives, telles que l'amniocentèse ou prélèvement de villosités choriales mai remplacer les procédures plus invasives telles que la prise de sang ftal ou biopsie de foie ftal pour le diagnostic prénatal de nombreuses maladies.
L'évaluation échographique des anomalies chromosomiques
Au cours des dix dernières années et demi, le domaine de l'échographie génétique a émergé. Il ya quinze ans l'amniocentèse était réservé aux femmes de l'âge maternel, d'où la possibilité d'identifier moins d'un quart des ftus aneuploïdie. Finalement, la décision de savoir si un patient est à risque d'aneuploïdie ftale sera basé sur la combinaison de l'âge maternel, les marqueurs biochimiques multiples, et une évaluation complète de l'échographie du ftus. Peut-être en utilisant cette méthode combinée, nous allons progresser depuis l'identification du potentiel de 20% des ftus atteints du syndrome de Down (ie, ceux associés à un âge maternel supérieur à 35 ans) à taux de détection possible de 90% des ftus présentant des anomalies chromosomiques, tout en recommandant amniocentèse pour seulement 5% à 10% des femmes enceintes.
Guidée par échographie obstétricale Diagnostic Procedures
Le syndrome de Down est l'anomalie chromosomique la plus fréquente chez les naissances vivantes et elle a démontré le syndrome le plus insaisissable de l'échographie tous les syndromes autosomique trisomique. Il semble logique, cependant, que l'examen échographique du ftus lui-même pour les caractéristiques reconnaissables de nourrissons atteints du syndrome de Down seraient encore plus sensibles et spécifiques pour la détection des ftus touchés que sont l'âge maternel et le dépistage sérique.
Échographiques de dépistage du syndrome de Down
Marqueurs échographiques d'aneuploïdie:
Deuxième trimestre d'examens échographiques sont souvent réalisé en routine pour détecter des anomalies anatomiques du foetus; les conclusions d'une anomalie majeure ou deux ou plusieurs mineurs malformations structurelles augmente la probabilité de l'aneuploïdie et les mandats de conseil génétique et l'examen de l'amniocentèse (7). Un certain nombre d'anomalies ftales structurelles et non-pathologiques (ou "soft") des marqueurs détectables à l'examen échographique de deuxième trimestre ont également été associées au syndrome de Down. Des études ont montré que l'examen de deuxième trimestre échographique peut identifier 50 à 75% des ftus atteints du syndrome de Down, à taux de dépistage positif de 4 à 15%. Toutefois, les performances des tests dépend de la population de l'étude, le choix des marqueurs, l'âge gestationnel et l'expertise de l'échographiste. Une approche courante consiste à utiliser les rapports de vraisemblance pour des marqueurs échographiques (calculé en divisant la sensibilité par le taux de faux positifs) afin de calculer le risque d'une femme d'avoir un foetus atteint du syndrome de Down; le risque fondée sur l'âge ou le dépistage sérique maternel est multiplié par le rapport de vraisemblance (8). Dans une étude prospective, population-based study cohorte impliquant des femmes avec des résultats du dépistage sérique qui indiquait un risque accru de syndrome de Down, la présence d'anomalies structurelles ou une nuque épaisse fois plus le risque de syndrome de Down par un facteur de neuf ans, et l'absence de anomalies réduit le risque d'un tiers. Lorsque les marqueurs mous ont été détectés dans le cadre d'une anomalie structurelle, le risque du syndrome de Down a en outre été augmenté, tandis que la constatation de marqueurs soft seul (sauf l'épaisseur nucale fois) n'ont pas modifié substantiellement le risque.
La signification des marqueurs soft détecté lors de l'examen échographique de routine à la gestation 20 semaines chez un patient qui se présente autrement d'être à faible risque de donner naissance à un enfant atteint de trisomie 21 ou trisomie 18 est incertain, et un sujet de préoccupation est que la présence de ces marqueurs mai conduire à des procédures invasives de diagnostic qui sont inutiles. Inversement, un examen échographique normal mai fournir un faux réconfort aux patients présentant un risque qui est augmenté sur la base de marqueurs sériques ou l'âge. Durant le premier trimestre, la non-visualisation de l'os nasal lors de l'examen échographique a été associée à un risque accru de syndrome de Down et d'autres aneuploïdies. Lorsqu'ils sont utilisés avec la projection du premier trimestre combinés chez les patients à risque accru en raison de l'âge maternel (âge médian: 35 ans), et le taux estimé de détection de la trisomie 21 a été 93% à un taux de dépistage positif de 5% (9). Évaluation de l'os nasal a été intégrée dans les algorithmes de dépistage dans certains centres, mais il devrait être effectuée uniquement par les échographistes formés.
Échographique conclusions dans la trisomie 21:
Structural anomalies: malformations cardiaques; hygromas kystique; atrésie duodénale; ventriculomégalie.
Constatations de non-pathologique, ou «marqueurs soft: épaisseur nucale fois (> 6 mm); hypoplasique ou absente os nasal; kystes du plexus choroïde; accent échogène intracardiaque; pyelectasis rénale; humérus court; fémur court; intestin échogène; cordon ombilical avec deux navires; hypoplasie de la phalange milieu du cinquième doigt; sandale-toe écart (séparation du gros orteil)
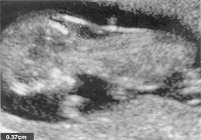
Augmentation de mesure de la clarté nucale de 3,7 mm à 12 semaines chez un foetus atteint du syndrome de Down
D'autres marqueurs du premier trimestre échographie objet d'une enquête (par exemple, conduit veineux au Doppler flux d'imagerie, insuffisance tricuspide foetale, et fronto-angles anormaux maxillaire et maxillaire de la mandibule) ne sont généralement pas utilisées dans les algorithmes de dépistage de routine dans la pratique (10). Pré-requis pour l'utilisation plus généralisée de ces mesures comprennent l'évaluation au sein des populations à faible risque, l'utilisation de données sur l'âge gestationnel et les normes qui sont spécifiques à la race du patient ou un groupe ethnique, l'adoption d'une approche normalisée, une formation adéquate, et la mise en uvre de la qualité programmes d'évaluation.
Résumé:
Ultrasound continuera à jouer un rôle crucial dans le développement de ces nouvelles approches de diagnostic prénatal du ftus et de thérapie. La disponibilité du premier trimestre de diagnostic prénatal précoce mai offrir des possibilités de soins avant la naissance de certains troubles par le remplacement des enzymes manquants ou cofacteurs, par remplacement génique somatique, ou par les transports en cellules souches. Coffre-fort et un accès fiable à la circulation ftale ouvre une nouvelle ère dans le diagnostic prénatal du ftus et de la thérapie ftale.
Références
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin. Invasive prenatal testing for aneuploidy. Obstet Gynecol 2007;110:1459-1467
- Nuaabaum RL, McInnes RR, Willard HF. Thompson & Thompson genetics in medicine. 7th ed. Philadelphia: Sunders/Elsevier, 2007
- The human genome. Science 2001;291:1145-1434
- Malone FD, D'Alton ME. First-trimester screening for Down syndrome. Obstet Gynecol 2003102:1066-1079
- Bahado-Singh RO, Oz UA, Mendilcioglu I et al. The mid-trimester genetic sonogram. Semin Perinatol 2005;29:209-214
- Simpson JL, Elias S. Genetics in obstetrics and gynecology. 3rd ed. Philadelphia, WB Saunders, 2003
- Alfirevic A, Sundberg K, Brigham S. Amniocentesis and chorionic villus sampling for prenatal diagnosis. Cochrane Database Syst Rev 2003;3:CD003252
- Smith-Bindman R, Chu P, Goldberg JD. Second trimester prenatal ultrasound for the detection of pregnancies at increased risk for Down syndrome. Prenat Diagn 2007;27:535-544
- Rosen T, D'Alton ME, Platt LD et al. First-trimester ultrasound assessment of the nasal bone to screen for aneuploidy. Obstet Gynecol 2007;110:399-404
- Driscoll DA, Gross S. Prenatal screening for aneuploidy. N Engl J Med 2009;360:2556-2562
Publié: 19 October 2009
Dedicated to Women's and Children's Well-being and Health Care Worldwide
www.womenshealthsection.com