


Évaluation du risque de cancer héréditaire en gynécologie
Bulletin WHEC pratique et directives cliniques de gestion pour les fournisseurs de soins de santé. Subvention à l'éducation fournie par la santé des femmes et Education Center (WHEC).
Évaluation du risque de cancer héréditaire devrait faire partie de l'obstétrique de routine et la pratique de la gynécologie. Au cours de la dernière décennie, il ya eu une prise de conscience accrue de la prédisposition héréditaire à une large gamme de maladies, dont le cancer, les maladies cardiaques et le diabète. Ce sont tous les troubles multi-géniques complexes, mais l'identification de gènes spécifiques associés à une prédisposition à ces conditions ont permis aux cliniciens d'évaluer plus précisément les risques et prescrivent des interventions préventives. Bien que cela puisse ne pas être familiers à de nombreux praticiens, le processus de stratification du risque de cancer peut être efficace. Utiliser évaluation protocole axé sur la susceptibilité au cancer, les facteurs de risque personnels et familiaux, et les tests génétiques, il est possible de créer des profils de risque et les stratégies de gestion qui démontrent une réduction prouvée dans la morbidité et la mortalité liées au cancer. Décennies avant que les tests génétiques entré pratique, les cliniciens avaient une charge excessive de cancer du sein et de l'ovaire, avec souvent des cas précoces et le cancer du sein chez l'homme. Dans le milieu des années 1990, l'analyse de liaison dans ces sein et des ovaires familles de cancer ont permis d'identifier des gènes BRCA1 et BRCA2 , des gènes qui fonctionnent normalement dans la réparation de l'ADN. conseillers en génétique du cancer utilisent l'analyse généalogique et les indicateurs cliniques pour évaluer la probabilité qu'un patient porteur d'une mutation héréditaire, afin de déterminer les stratégies de test appropriées parmi un éventail de plus en plus complexe de tests disponibles dans le commerce, et d'interpréter les résultats et d'orienter les recommandations médicales. La découverte scientifique, la sensibilisation du public répandue et la disponibilité générale de tests génétiques cliniques du BRCA1 et BRCA2 gènes responsables de la majorité des cancers du sein familiaux et les cancers de l'ovaire, ont conduit les femmes à présenter aux bureaux de médecins demandant des tests génétiques et remettre en question les implications cliniques pour eux et leurs familles.
Le but de ce document pour examiner les recommandations actuelles pour le dépistage génétique de la susceptibilité aux cancers, y compris les ovaires, des trompes de Fallope, du sein, de l'endomètre et du côlon dues à des mutations héréditaires dans les gènes BRCA gènes ou dans les gènes de réparation des mésappariements associés au cancer du côlon héréditaire sans polypose (HNPCC) syndrome. L'histoire familiale demeure la pierre angulaire de l'identité du patient. Ethnicité et la race sont systématiquement évalués dans le cadre de l'évaluation. Les applications pratiques de dépistage génétique de prédisposition au cancer ont la capacité de réduire le fardeau des cancers héréditaires en sauvant des vies, diminuer morbidités médicales, et réduire le stress psychologique. Gestion des porteurs de la mutation, y compris les indications pour les chirurgies de réduction des risques, le dépistage du cancer et de suivi, sont également abordées. L'examen porte également sur les pratiques cliniques et l'utilisation pratique de la génétique clinique sur le cancer.
Contexte
Il existe de nombreux syndromes qui confèrent et hérités risque de cancer du sein, de l'ovaire et les cancers liés avec 5% à 15% de tous les cancers du sein et de l'ovaire causé par un de ces syndromes, de cancer héréditaire dominante autosomique (1),(2). Les gènes associés à des syndromes de cancers héréditaires sont tous transmis dans un mode autosomique dominant mendélienne, les antécédents familiaux de sorte maternels et paternels contribuent à part égale au risque du patient et doivent être évalués pour déterminer la pertinence des tests. Il convient de noter, toutefois, que, bien que les mutations génétiques sont héritées dans un mode autosomique dominant, elles sont exprimées de manière récessive (2). Une personne qui hérite d'une mutation de lignée germinale qui inactive une copie du gène a en général une deuxième copie intacte du gène fonctionnel. Ce n'est que lorsque cette copie normale restant devient muté que la cellule peut subir le processus de transformation maligne.
Le rôle des obstétriciens et gynécologues implique:
- Reconnaissant l'évolution des maladies familiaux évocateurs d'une prédisposition héréditaire au cancer, y compris les syndromes familières et courantes de cancer héréditaire du sein et le cancer de l'ovaire (HBOC) et héréditaire sans polypose cancer colorectal (HNPCC ou syndrome de Lynch);
- Intégrer l'évaluation des risques, les tests génétiques et l'interprétation des résultats dans la pratique quotidienne;
- Orienter la gestion médicale basée sur la stratification du risque.
stratification du risque: Risque sporadique, Risque familial, et risque héréditaire
Plus de 10% des patients ont des antécédents de santé personnels ou familiaux suggérant susceptibilité au cancer héréditaire ou familiale, et plus de 6% des patients répondent aux critères National Comprehensive Cancer Network (NCCN) pour les tests génétiques (3). Trois profils de risque ont émergé:
- Risque sporadique, définie comme la population moyenne ou le patient à faible risque;
- Risque familial, défini comme une famille ayant de nombreux parents avec un type ou un cancer spécifique;
- Risque héréditaire, définie comme la présence d'un seul cancer ou d'un syndrome de tumeurs malignes dans une famille, qui sont associées à des mutations délétères héréditaires connus de gènes spécifiques (par exemple, BRCA ).
Risque héréditaire porte le pourcentage le plus élevé de susceptibilité au cancer, tandis que le risque sporadique porte le plus bas. Avec la stratification du risque, nous pouvons identifier les personnes qui pourraient bénéficier d'un dépistage intensif, les tests génétiques et les interventions telles que la chimioprévention et la réduction du risque chirurgical. Les tests génétiques de personnes concernées nous permet en outre d'identifier les patients atteints de syndromes héréditaires de cancer, pour leur propre bénéfice et celui de toute leur famille. Une fois une histoire familiale de cancer est identifié, les modèles décrits ci-dessous sont utilisés pour prédire un cancer particulier et la probabilité d'une mutation génétique qui prédispose le patient à un syndrome de cancer héréditaire. Le consentement éclairé, y compris les risques, les avantages, les options et les attentes, devrait être examiné de manière adéquate. Conseil direct est nécessaire dans le cas d'une famille anormale ou l'histoire de cancer personnelle. Consultation non directe réduit la possibilité pour le patient pour une surveillance accrue et un potentiel diagnostic précoce et la prévention du cancer, et met le médecin à risque de responsabilité future.
Cancer héréditaire du sein et le cancer (HBOC) Le syndrome des ovaires
Cancer héréditaire du sein et le cancer (HBOC) syndrome de l'ovaire est un syndrome du cancer prédisposition héréditaire. Les caractéristiques de ce syndrome sont multiples membres de la famille atteints de cancer du sein ou un cancer de l'ovaire ou des deux, la présence à la fois le cancer du sein et le cancer de l'ovaire chez un seul individu et l'âge précoce de survenue de cancer du sein. Au milieu des années 1990, il a été démontré que hérité des mutations dans le gène BRCA1 et BRCA2 gènes sur les chromosomes 17 et 13, respectivement, étaient responsables de la poitrine la plus familiale et les cancers de l'ovaire. Les femmes et les hommes peuvent porter un mutant BRCA gène et de le transmettre à leur progéniture.
«Drapeaux rouges» pour le syndrome HBOC - histoire personnelle et familiale de trois générations, y compris (4):
- Cancer du sein: préménopause ou sous l'âge de 50 ans, bilatérale, triple négatif, ou un homme;
- Cancer de l'ovaire: n'importe quel âge, généralement épithéliale, de haute qualité séreux;
- Le cancer du pancréas, le mélanome ou cancer de la prostate: de moins de 50 ans;
- Prédisposition ethnique: juive ashkénaze et d'autres (par exemple, mexicaine, islandais, néerlandais, hongrois);
- A connu BRCA mutation dans la famille.
BRCA1 et BRCA2
Environ 10% des cas de cancer de l'ovaire et 3-5% des cas de cancer du sein sont dus à des mutations germinales dans les gènes BRCA1 et BRCA2 (1),(5). BRCA1 se trouve sur le chromosome 17, et BRCA2 est situé sur le chromosome 13. Plus de 1200 mutations différentes ont été rapportées pour BRCA1 , et plus de 1300 mutations différentes ont été rapportées pour BRCA2 . BRCA1 et BRCA2 sont des gènes suppresseurs de tumeur qui codent pour des protéines qui fonctionnent dans le processus de réparation de l'ADN (6). Bien que les personnes atteintes du syndrome HBOC héritent d'un allèle défectueux dans BRCA1 et BRCA2 de leur père ou de la mère, ils ont une deuxième allèle fonctionnel. Si le second allèle devient non fonctionnel, le cancer peut se développer à travers l'accumulation de mutations supplémentaires. C'est ce qu'on appelle l '«hypothèse de deux hit" (7). Dans la population générale, on estime que près de 1 sur 300 à 1 800 individus porteurs d'une mutation dans le gène BRCA1 et BRCA2 (8). La population juive ashkénaze représente une exception notable, car il ya trois mutations fondatrices spécifiques (BRCA1 codon 185 deletion AG, BRCA1 codon 5374 insert C, et BRCA2 codon 6174 suppression T) qui sont transportées par environ 2,5% des individus (8). Dans cette perspective, la plupart des experts ont un seuil beaucoup plus bas pour les tests génétiques chez les femmes juives.
Pour une femme, avec une BRCA1 mutation, le risque de cancer de l'ovaire est 39-46%. Pour une femme avec un BRCA2 mutation, le risque de cancer de l'ovaire est 12-20%. Le risque à vie estimé de cancer du sein avec un BRCA1 ou BRCA2 mutation est 65-70% (9) . Pour les femmes atteintes d'un cancer du sein, le risque actuariel de 10 ans de développer un cancer de l'ovaire subséquente est de 12,7% pour BRCA1 porteurs de la mutation et de 6,8% pour BRCA2 porteurs de la mutation (10). Cancer de l'ovaire associé à BRCA1 et BRCA2 mutations a un phénotype histologique distincte. Ce type de cancer est principalement de l'histologie séreuse ou endométrioïde et de haute qualité. Cancers de l'ovaire et mucineuses borderline ne semblent pas faire partie du spectre tumeur (11). Primaire cancer des trompes de Fallope et le cancer péritonéal primaire sont également du spectre de la maladie associée à BRCA1 et BRCA2 mutations (12).
Critères d'évaluation du risque génétique
Les patients atteints supérieure à une participation d'environ 20-25% de chance d'avoir une prédisposition héréditaire au cancer du sein et le cancer de l'ovaire et pour qui l'évaluation du risque génétique est recommandé (13):
- Les femmes ayant des antécédents personnels de cancer du sein à la fois et le cancer ovarien*
- Les femmes atteintes d'un cancer ovarien* et un proche parent atteints de cancer de l'ovaire ou un cancer du sein avant la ménopause ou les deux;
- Les femmes atteintes d'un cancer ovarien* qui sont d'ascendance juive ashkénaze;
- Les femmes atteintes d'un cancer du sein à l'âge de 50 ans ou moins et un proche parent atteintes d'un cancer ovarien* ou un cancer du sein chez l'homme à tout âge;
- Femmes du ascendance juive ashkénaze chez qui un cancer du sein a été diagnostiqué à l'âge de 40 ans ou moins;
- Les femmes ayant un proche parent avec un connu BRCA1 ou BRCA2 mutation.
*Le cancer du péritoine et de tubes de Fallope doit être considérée comme une partie du spectre du syndrome HBOC.
proche parent est défini comme un parent au premier degré (mère, sur, fille) ou du deuxième degré (mère, fille, tante, nièce).
Les patients atteints supérieur à un 5-10% de chances approximative d'avoir une prédisposition génétique au cancer du sein et le cancer de l'ovaire et pour qui l'évaluation du risque génétique peut être utile (13):
- Les femmes atteintes d'un cancer du sein à l'âge de 40 ans ou moins;
- Les femmes atteintes d'un cancer de l'ovaire, cancer péritonéal primaire, ou un cancer des trompes de Fallope de haute qualité, l'histologie séreuse à tout âge;
- Les femmes atteintes d'un cancer du sein bilatéral (en particulier si le premier cas de cancer du sein a été diagnostiqué à l'âge de 50 ans ou moins);
- Les femmes atteintes d'un cancer du sein à l'âge de 50 ans ou moins et un proche parent d'un cancer du sein à l'âge de 50 ans ou moins;
- Femmes du ascendance juive ashkénaze avec le cancer du sein à l'âge de 50 ans ou moins;
- Les femmes atteintes d'un cancer du sein à tout âge et deux ou plus proches avec le cancer du sein à tout âge (surtout si au moins un cas de cancer du sein a été diagnostiqué à l'âge de 50 ans ou moins);
- Les femmes ne sont pas touchés par un proche parent qui répond à un des critères précédents.
Outil d'évaluation du risque de syndrome HBOC
Nous avons développé un outil simple d'enseignement numérique qui peut être utilisé pour estimer la candidature d'un patient pour BRCA tests. Cet outil se rapproche directives du NCCN en attribuant 1 ou 2 points à chaque personne avec chaque «drapeau rouge» cancer pertinent dans l'arbre généalogique sur trois générations. Les points de l'patients sont additionnés avec les points maternelle, puis de nouveau avec les points paternels. Une somme de point 0 indique une classification sporadique (faible). Une somme de 1 point devrait normalement indiquer une classification de risque familial (moyen), mais un patient 1 point peut encore se qualifier pour les tests génétiques s'il ya une structure familiale limitée ou une prédisposition ethnique BRCA mutations. Une somme de> 2 points sera généralement qualifier pour BRCA tests, bien que les combinaisons qui impliquent des parents au troisième degré peuvent être évaluées afin de déterminer si le test est justifiée. Cet outil ne doit être utilisé comme une estimation, et non un guide d'essais concluants.
Entourez chaque point de cancer qui s'applique, alors totale a+b et a+c
- Tout 1 point = risque familial; possible risque héréditaire si petite famille (LFS)
- Tous les 2 points = BRCA candidat de test
Chaque cancer primaire compte séparément
Famille = 1er relations familiales degré et Les relations familiales 2e degré (
les relations familiales parfois 3e degré)
Scores maternelle et paternelle ajouter séparément pour les patients
| Patient (a) | Maternal (b) | Paternal (c) | |
| Breast, <50 years (premenopause) | 2 | 1 | 1 |
| Breast, <60 years, triple negative | 2 | 1 | 1 |
| Breast, >50 years, not triple negative | 1 | 1 | 1 |
| Breast, bilateral, any age | 2 | 2 | 2 |
| Breast, male, any age | 2 | 2 | 2 |
| Ovary, epithelial, any age | 2 | 2 | 2 |
| Pancreas | 1 | 1 | 1 |
| Known mutation carrier | - | 2 | 2 |
| Ashkenazi Jewish (or other high-risk group) |
1 | 0 | 0 |
| Total: a+b = ---------- points; a+c = ---------- points | |||
HBOC, hereditary breast and ovarian cancer; LFS, Li-Fraumeni syndrome
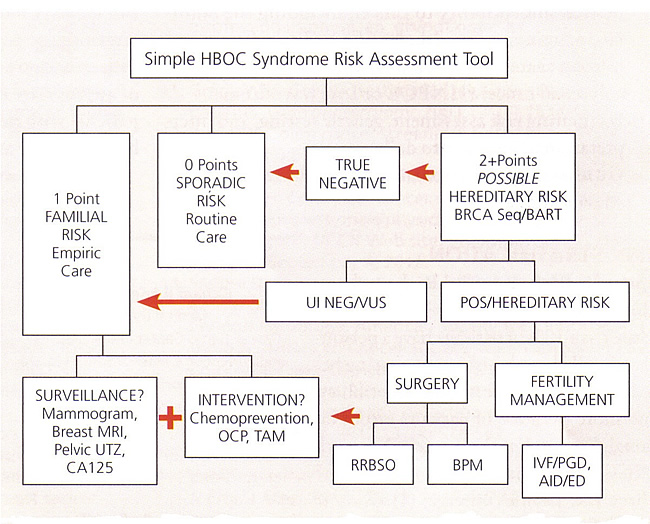
Abbreviations: AID, artificial insemination by donor; BART, BRCA Analysis rearrangement test; BPM, bilateral prophylactic mastectomy; CA 125, cancer antigen 125; ED, egg donation; HBOC, hereditary breast and ovarian cancer; IVF, in-vitro fertilization; MRI, magnetic resonance imaging; OCP, oral contraceptive pills; PGD, preimplantation genetic diagnosis; POS, positive; RRBSO, risk-reducing bilateral salpingo-oophorectomy; TAM, tamoxifen; UI NEG, uninformative negative; UTZ, ultrasound; VUS, variant of uncertain significance.
Le conseil génétique pour réduire le risque de cancer du sein
Le conseil génétique devrait inclure une discussion des résultats possibles de tests - portant spécifiquement sur les questions de résultats positifs, négatifs et peu informatives, ou des variantes de signification inconnue. Options pour la surveillance, la chimioprévention et la chirurgie de réduction des risques doivent être discutées avant le test. Conséquences psychologiques et familiaux possible des résultats des tests devraient aussi être considérés. Les documents écrits peuvent aider les gens à partager l'information avec les parents au sujet de leurs risques génétiques potentiels. Le conseil génétique devrait également inclure une discussion sur le coût des tests génétiques. Plusieurs compagnies d'assurance, y compris Medicare, couvrent une partie importante de la dépense pour certaines personnes. Medicare et d'autres compagnies d'assurance ont écrit des lignes directrices pour couvrir le coût des tests génétiques. Un aspect important du conseil génétique est la discussion de la législation actuelle en matière de discrimination génétique et la confidentialité des renseignements génétiques. The Genetic loi fédérale de non-discrimination de l'information de 2008 protège les individus contre la santé et de la discrimination dans l'emploi fondée sur l'information génétique. De nombreux États ont également des lois de l'État qui offrent une protection similaire. Ces lois ne s'appliquent pas à d'autres formes d'assurance, qui peuvent inclure l'assurance vie ou invalidité.
Idéalement, il est logique de commencer à expérimenter dans un individu affecté. Si, toutefois, aucune personne touchée est disponible, les tests génétiques peuvent encore fournir des informations utiles. Si une mutation délétère est identifiée, les patients à risque peuvent être conseillés de manière appropriée sur le dépistage ou d'autres approches de réduction des risques. Si aucune mutation délétère est identifiée, les patients doivent être avisés que cela pourrait être dû à l'une des nombreuses possibilités, y compris (a) une mutation délétère présent dans la famille que le patient n'a pas hérité; (b) une mutation indétectable dans les gènes BRCA1 et BRCA2 , ou un gène de susceptibilité au cancer non identifié est pourtant présent dans la famille, et on ne sait pas si le patient partagent cette prédisposition, ou (c) aucune prédisposition héréditaire dans la famille.
Les stratégies actuelles pour réduire le risque de développer un cancer de l'ovaire ou un cancer des trompes de Fallope chez les femmes à haut risque avec délétères connus BRCA mutations comprennent la surveillance, la chimioprévention et la chirurgie. Compte tenu du risque très élevé de cancer de l'ovaire et le cancer des trompes de Fallope chez les femmes avec des mutations dans BRCA1 et BRCA2 , groupes de concertation ont recommandé un dépistage périodique avec CA 125 et l'échographie transvaginale, à commencer entre les âges de 30 ans et 35 ans ou 5-10 ans plus tôt que le plus jeune âge du premier diagnostic de cancer de l'ovaire dans la famille (14). La faible prévalence du cancer de l'ovaire et de la forte probabilité d'un résultat de test de dépistage positif qui nécessite une évaluation plus poussée chirurgicale invasive des obstacles dans les programmes de dépistage du cancer de l'ovaire chez les femmes au risque héréditaire (15). Il est à espérer que le futur procès de cancer de l'ovaire bénéficiera de meilleurs marqueurs sériques et les algorithmes de dépistage améliorées pour accroître la capacité de distinguer entre CA 125 valeurs normales et anormales et les résultats de l'échographie. Les avantages et l'importance de la réduction du risque des contraceptifs oraux pour les femmes avec un BRCA mutation n'a pas été rapporté de manière aussi cohérente que pour la population en général, à faible risque. Il est raisonnable pour les femmes avec des mutations dans BRCA1 et BRCA2 , d'utiliser des contraceptifs oraux. La plupart des études rapportent une diminution du risque de cancer de l'ovaire chez ceux qui ont utilisé des contraceptifs oraux pour une durée plus longue (plus de 3 à 6 ans) (16) . Les risques et les avantages relatifs à la fois pour la chimioprévention et le contrôle de la reproduction doivent être soigneusement pesés par le patient et son médecin.
Prophylactique salpingo-ovariectomie bilatérale (BSO)
Il est fortement recommandé chez les femmes porteuses du gène BRCA mutations en raison du taux élevé de mortalité du cancer de l'ovaire et de l'absence de contrôle efficace et les méthodes de prévention (17). Heureusement, le risque de cancer héréditaire du sein et le cancer de l'ovaire ne pas augmenter de façon spectaculaire jusqu'à la fin des années 30 chez les femmes atteintes BRCA1 mutations, et la fin des années 50 pour les femmes avec BRCA2 mutations (18), de sorte que les femmes aient la possibilité de compléter leurs familles avant de subir BSO. La pratique antérieure de réaliser une chirurgie prophylactique basée uniquement sur l'histoire de la famille devrait être en grande partie abandonné. La moitié des femmes dans les familles avec BRCA ne serait pas prévu mutations à être porteurs. Actuellement, la décision de procéder à une chirurgie prophylactique BSO se fonde principalement sur les résultats de BRCA analyse mutationnelle. Il existe certaines preuves que certains types de gènes BRCA mutations prédisposent davantage au cancer de l'ovaire, mais cette observation n'a pas été toujours confirmé (19). Bien BSO est une procédure chirurgicale majeure, des études montrent que la plupart des femmes à haut risque subissant des tests génétiques acceptent salpingo-ovariectomie bilatérale. Laparoscopie et la laparotomie sont à la fois des options pour réduction des risques BSO. Pour les deux procédures, une inspection approfondie des surfaces péritonéales est nécessaire. Lavage péritonéal doivent être obtenues. Le diaphragme, le foie, épiploon, l'intestin, les gouttières pariéto-coliques, et l'annexe sont inspectés dans l'abdomen. Les ovaires, les trompes de Fallope, l'utérus, de la vessie, séreuse et cul-de-sac sont inspectés dans le bassin. Toutes les zones anormales devraient subir une biopsie. Les vaisseaux ovariens doivent être isolés et liés proximale à l'extrémité du tissu ovarien identifiable afin de s'assurer que tous les tissus de l'ovaire est complètement enlevé. Si une hystérectomie n'est pas effectuée, la trompe de Fallope devrait être divisée à son insertion dans la corne utérine. Lorsque vous effectuez une procédure laparoscopique, pour optimiser la préservation de l'épithélium de surface de l'ovaire, les échantillons peuvent être placés dans un sac endoscopique avant le retrait de l'abdomen. La décision de pratiquer une hystérectomie concomitante doit être individualisée. Les arguments en faveur de l'hystérectomie comprennent une stratégie de thérapie hormonale plus simplifiée (avec de l'oestrogène seulement) et un risque accru de cancer théorique dans la trompe de Fallope cornée (20). En outre, l'hystérectomie peut être envisagée lorsqu'il ya d'autres indications médicales pour ablation de l'utérus et du col utérin. Pour les femmes prenant du tamoxifène, l'hystérectomie peut être envisagée pour réduire leur risque de cancer de l'endomètre (21).
Avantages et inconvénients de la réduction des risques Salpingo ovariectomie
Avantages:
- Diminue l'ovaire et de Fallope incidence du cancer du tube et la mortalité;
- Peut souvent être retardé pour permettre l'achèvement de procréer;
- Approche laparoscopique possible dans la plupart des cas;
- Impact de l'image du corps généralement acceptable;
- Remplacement de l'oestrogène peut prévenir les conséquences de la ménopause chirurgicale;
- Diminue le risque de cancer du sein.
Inconvénients
- coût;
- Morbidité et la mortalité potentielle;
- Petit mais résiduel potentiel ultérieur cancer péritonéal primaire;
- La ménopause chirurgicale chez les patientes préménopausées qui choisissent de ne pas prendre hormonal substitutif.
Quel âge à prendre en considération à réduire les risques Salpingo ovariectomie?
Réduction des risques et élective salpingo-ovariectomie est l'ablation des ovaires et des trompes de Fallope pour le bénéfice potentiel de prévention de la morbidité et de la mortalité à long terme. Le terme de réduction des risques ovariectomie implique que les ovaires sont normaux au moment de l'enlèvement. Les femmes ayant BRCA1 ou BRCA2 mutations devraient être offerts de réduction des risques ovariectomie par l'âge de 40 ans, ou lorsque la procréation est complet (13). Cancer de l'ovaire seront diagnostiqués en moins de 2-3% des femmes atteintes de BRCA1 ou BRCA2 mutations avant l'âge de 40 ans. Pour les femmes avec BRCA1 mutations, le risque de cancer de l'ovaire augmente façon marquée au cours des années 40, avec 10-21% de BRCA1 porteurs de la mutation de développer un cancer de l'ovaire par l'âge de 50 ans. Le risque de cancer de l'ovaire avant la ménopause est beaucoup plus faible dans BRCA2 porteurs de la mutation, avec pas plus de 3% de BRCA2 porteurs de la mutation de développer un cancer de l'ovaire par âge de 50 ans (13),(22). Compte tenu de la modification du calendrier du risque de cancer de l'ovaire, l'examen peut être faite pour conseiller les patients avec BRCA1 mutations différemment que pour BRCA2 porteurs de la mutation. Cependant, les femmes ayant BRCA2 mutations ont une chance de 26-34% de développer un cancer du sein de 50 ans (18),(22), et le bénéfice maximum de l'ablation des ovaires sur la réduction du risque de cancer du sein est réalisé le plus tôt les ovaires sont enlevés (23). Compte tenu de ces enjeux, le calendrier de réduction des risques ovariectomie doit être basée sur les besoins de chaque patient, en tenant compte de leur désir de préserver la fertilité ou d'empêcher prématurée ménopause chirurgicale à l'impact dépend de l'âge de la réduction des risques ovariectomie sur les deux seins cancer et les risques de cancer gynécologique.
Salpingectomie prophylactique et ovariectomie retardée
Prophylactique salpingo-ovariectomie bilatérale est conseillé pour les femmes BRCA mutations, mais il ya des conséquences néfastes de la ménopause prématurée. La majorité des BRCA cancers de l'ovaire associés semblent survenir dans la trompe de Fallope, par conséquent, salpingectomie peut être une alternative à salpingo-ovariectomie bilatérale. Avec la compréhension que peut-être 60% des cancers séreux pelviens peut provenir de la trompe de Fallope, il ya eu une discussion plus récente de réduction des risques salpingectomy comme une alternative potentielle au risque de réduction de BSO pour le traitement de BRCA porteurs de la mutation, en particulier chez les femmes réticentes à subir de réduction des risques BSO (39). Les auteurs publient leur Markov Monte Carlo comparant BSO de réduction du risque à 40 ans avec deux salpingectomy de réduction du risque à 40 ans suivis par une ovariectomie bilatérale retardée à 50 ans (40). Ce modèle réfléchie prédit la baisse attendue des cancers pelviens séreuses, les cancers du sein et de décès supplémentaires dus aux maladies cardio-vasculaires. La conclusion de l'étude était salpingo-ovariectomie bilatérale souvent le plus grand de la réduction des risques de cancer du sein et de l'ovaire chez les porteuses de mutations BRCA. Toutefois, lorsque l'on considère l'espérance de vie ajustée sur la qualité, la salpingectomie bilatérale avec ovariectomie différée est une stratégie rentable et peut être une alternative acceptable pour ceux qui refusent de se soumettre à salpingo-ovariectomie bilatérale (40). Il est important de souligner que la norme de soins pour les femmes qui héritent des mutations germinales dans les gènes BRCA1 et BRCA2 reste BSO prophylactique après l'achèvement de procréer ou vers l'âge de 40 ans. Il offre la plus grande réduction des risques dans les cancers du sein et de l'ovaire par rapport à salpingectomy avec ou sans ovariectomie retardée. Toutefois, une proportion importante de femmes ne subissent pas BSO, et beaucoup choisissent surveillance seul pour le cancer de l'ovaire, malgré l'avantage limité de méthodes de dépistage existants (41). Le cancer des ovaires entraîne le taux de mortalité chez BRCA porteurs de la mutation, la donc toute intervention qui réduit le risque de cancer de l'ovaire est susceptible d'être validé de façon prospective, salpingectomie bilatérale avec ovariectomie retardé peut être une alternative raisonnable à BSO, surtout pour ceux qui sont réticents à subir ce dernier procédure en raison de l'effet potentiel sur la qualité de vie.
Gestion des femmes avec des antécédents familiaux et négatif BRCA1 ou BRCA2 Mutation
Bien que, dans la plupart des cas, une prédisposition génétique au cancer de l'ovaire est causée par des mutations BRCA1 ou BRCA2 , la technologie actuelle ne permet pas d'identifier toutes les mutations qui doivent exister dans ces gènes (24). En outre, les liens études ont suggéré que c'est moins de la moitié des familles de quatre ou plus de cas de cancer du sein, mais aucun cas de cancer de l'ovaire (familles avec un site spécifique du cancer du sein), le cancer du sein sont causés par BRCA1 ou BRCA2 mutation (25). Compte tenu de ces problèmes, les femmes ayant des antécédents personnels ou familiaux de cancer du sein qui ont testé négatif pour un BRCA mutation doivent être gérées en fonction de leur histoire familiale. Les données préliminaires ont suggéré que les femmes issues de familles ayant des antécédents de cancer du sein spécifique au site dans lequel aucun BRCA mutation est identifiée restent à un risque significativement accru de cancer du sein, mais peut-être pas à une augmentation significative du risque de cancer de l'ovaire (26). Il est important pour les individus à haut risque de rester en contact avec des cliniciens expérimentés dans la prise en charge des femmes à risque accru, étant donné le développement rapide de la recherche et des améliorations dans la technologie de test. Par exemple, un test pour les grands réarrangements dans les gènes BRCA1 et BRCA2 gènes a été développé qui peut aider à identifier des mutations dans un petit pourcentage des familles à risque élevé qui ont déjà été testés négatifs pour ces gènes.
Options de reproduction
Les résultats des tests génétiques peuvent avoir un impact profond sur les décisions de planification familiale pour les personnes en âge de procréer qui sont jugées porteuses de BRCA1/2 mutations. Par exemple, dans le cas où les deux partenaires exercent une BRCA2 mutation, il peut y avoir un risque élevé pour la progéniture de développer une rare anémie de Fanconi / cerveau phénotype tumoral (maladie récessive). Counseling pour les options de reproduction tels que le diagnostic prénatal, le diagnostic préimplantatoire (DPI) et la procréation assistée peut donc être justifiée pour les couples exprimant des inquiétudes sur le BRCA mutation statut de porteur de leur future progéniture. L'orientation devrait comprendre une discussion approfondie sur les risques potentiels, les avantages et les limites des options de reproduction. Le diagnostic prénatal implique une analyse génétique post-implantation de l'embryon précoce, en utilisant des villosités choriales ou des échantillons de cellules du liquide amniotique; tests génétiques est généralement effectuée entre la semaine 12 et la semaine 16 de gestation, et les résultats des tests peuvent potentiellement conduire à la décision d'un couple d'interrompre la grossesse (27).
Au cours des deux dernières décennies, le DPI a émergé comme une méthode alternative aux tests génétiques dans les embryons précoces. DPI implique le test de 1 ou 2 cellules provenant d'embryons à un stade très précoce de développement (par exemple, 6 à 8 cellules) après fécondation in vitro (FIV). Cette procédure permet de sélectionner des embryons sains pour être transférés dans l'utérus, et peut donc offrir l'avantage d'éviter la cessation potentiel de grossesse. Toutefois, les procédures telles que le DPI ne sont pas sans limites car il peut toujours exiger un diagnostic prénatal de confirmation selon les besoins ou des demandes médicales d'un couple. En outre, le processus DPI nécessite l'utilisation de la FIV indépendamment de l'état de fertilité du couple (par exemple, s'applique également aux couples sans problèmes d'infertilité), et la FIV peut pas toujours mener à une grossesse réussie. Enfin, la technologie ou le savoir-faire ne peuvent pas être facilement accessibles dans la situation géographique d'un couple. Divers facteurs, à la fois médicaux et personnels, doivent être pesés dans la décision d'utiliser un diagnostic prénatal ou DPI. Considérations d'ordre médical peuvent inclure des facteurs tels que l'âge d'apparition de la tumeur, la pénétrance, la gravité ou associé morbidité et de mortalité des cancers héréditaires, et la disponibilité des méthodes de réduction des risques efficaces contre le cancer ou un traitement efficace. Bien que l'utilisation du diagnostic prénatal ou DPI est relativement bien établie pour les troubles héréditaires graves avec une très grande pénétrance, leur utilisation dans des conditions associées à pénétrance inférieur (par exemple, cancer héréditaire du sein ou le syndrome du cancer de l'ovaire) reste quelque peu controversé d'un point de vue éthique et réglementaire. Considérations personnelles de la décision d'utiliser un diagnostic prénatal ou DPI peuvent inclure des croyances individuelles éthiques, les systèmes de valeurs, les croyances culturelles et religieuses, ainsi que des facteurs sociaux et économiques. Basé sur les résultats de sondages administrés aux femmes à haut risque de cancer héréditaire du sein ou un cancer de l'ovaire, 50% -75% des répondants estiment que le DPI était une option acceptable pour personne à risque élevé encore que d'environ 14% -33% envisagent avoir PGD eux-mêmes (28). Surtout, les enquêtes suggéré que la majorité des femmes à haut risque ont peu ou aucune connaissance du DPI, en soulignant la nécessité d'une meilleure sensibilisation et d'éducation concernant les options en matière de reproduction potentiels. Naissances réussies ont été rapportés avec l'utilisation du DPI et de la FIV dans BRCA1/2 porteuses d'une mutation, mais les données de la littérature sont encore très limitées. En outre, les données relatives à la sécurité ou les résultats de reproduction PDG et assisté à long terme du gène BRCA porteurs de la mutation ne sont pas encore disponibles.
Héréditaire du cancer du côlon de polypose (HNPCC) ou syndrome de Lynch
Il s'agit d'une maladie héréditaire autosomique dominant du système de réparation des mésappariements de l'ADN. De nombreuses autorités se réfèrent à HNPCC comme le syndrome de Lynch, estimant que HNPCC ne reconnaît pas suffisamment l'importance de tumeurs malignes extracoliques, comme l'endomètre, de l'ovaire, tractus gastro-intestinal supérieur, et les cancers des voies urinaires associés à cette maladie. Ce syndrome représente 5-10% de l'ensemble du côlon et de l'endomètre (29). L'analyse de liaison des familles à risque élevé ont conduit à la découverte que le syndrome de Lynch est causée par des mutations germinales dans une classe de gènes responsables de la réparation de certains types de mutations de l'ADN (29). Ces gènes sont appelés gènes "de réparation des mésappariements" et donnent lieu à des protéines qui relisent l'ADN et de corriger les erreurs qui sont faites au cours du processus normal de réplication. MSH2 (MutS homologue 2) et MLH1 (MutL L homologue 1) sont le décalage le plus souvent muté réparer des gènes dans ce syndrome. MSH2 et MLH1 sont situés sur les chromosomes 3p21 et 2P16, respectivement. Mutations germinales dans d'autres gènes de réparation des mésappariements (MSH6 , PMS1 et PMS2 ) ont été identifiés, mais à une fréquence inférieure (29).
Probabilité de développer un cancer si il est sur la lignée germinale MLH1 ou MSH2 Mutation:
| Cancer Type | Risk at Age 70 (%) |
| Colorectal carcinoma | 80 |
| Endometrial carcinoma | 42 |
| Gastric carcinoma | 19 |
| Biliary tract carcinoma | 18 |
| Urinary tract carcinoma | 10 |
| Ovarian carcinoma | 10 |
L'histoire familiale est la première étape pour déterminer si un patient est à risque accru pour une maladie héréditaire. Plusieurs critères relativement stricts ont été développés pour identifier les personnes à risque élevé de syndrome de Lynch. Environ 30% des familles répondant aux critères de Bethesda et 50-92% des familles remplissant les critères d'Amsterdam auront ADN de lignée germinale inadéquation mutation de gène de réparation (30),(31).
Les critères de Bethesda pour HNPCC (voir ci-dessous) semblent être les plus sensibles à la prévision des mutations de réparation des mésappariements dans les familles HNPCC (30). Tous les éléments suivants:
- Carcinome colorectal moins de 50 ans;
- Présence des carcinomes HNPCC liés synchrones ou métachrones, indépendamment de l'âge (colorectal, de l'endomètre, de l'estomac, des ovaires, du pancréas, de l'uretère, bassinet du rein, des voies biliaires, la glande sébacée et l'intestin grêle);
- Carcinome colorectal avec des caractéristiques histologiques spécifiques à un individu de moins de 60 ans d'âge. Caractéristiques: lymphocytes infiltrant les tumeurs, Crohn's réaction de type lymphocyte, mucinous / Chevalière différenciation, ou modèle de croissance médullaire;
- Carcinome colorectal diagnostiqué chez un ou plusieurs parents de premier degré-colorectaux avec HNPCC liés à l'un des diagnostics effectués sous l'âge de 50 ans;
- Carcinome colorectal dans deux ou plus de première ou de deuxième degré avec une tumeur HNPCC liés, indépendamment de l'âge.
Les critères de Amsterdam II (voir ci-dessous) pour HNPCC sont plus spécifiques (31). Tout ce qui suit:
- Carcinome colorectal et / ou de carcinome de l'endomètre ou de carcinome à cellules transitionnelles de l'uretère ou bassinet du rein ou de cancer de l'intestin grêle dans au moins trois personnes de la même famille;
- Un des patients est un parent au premier degré de deux autres patients;
- La maladie survient dans au moins deux autres membres de la famille;
- Au moins un des diagnostics a été faite avant l'âge de 50 ans;
- Le diagnostic doit être confirmé histologiquement;
- La polypose adénomateuse familiale est exclue.
HNPCC / Lynch Syndrome de l'outil d'évaluation simple des risques
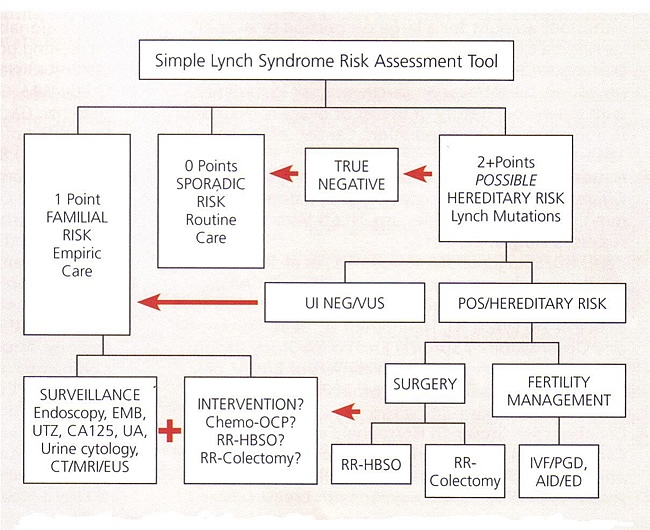
Abbreviations: AID, artificial insemination by donor; CA 125, cancer antigen 125; CT, computed tomography, ED, egg donation; EMB, endometrial biopsy; EUS, endoscopic ultrasound; HBSO, hysterectomy with bilateral salpingo-oophorectomy; IVF, in-vitro fertilization; MRI, magnetic resonance imaging; OCP, oral contraceptive pills; PGD, preimplantation genetic diagnosis; POS, positive; RR, risk-reducing; UA, urinalysis; UI NEG, uninformative negative; UTZ, ultrasound; VUS, variant of uncertain significance.
La surveillance des cancers héréditaires sans polypose du colon (HNPCC)
Lorsque les mutations sont identifiées, une analyse plus simple, moins coûteux, génétique peut être effectuée sur d'autres membres de la famille. Vaste consultation est recommandée avant et après le test. Une fois le processus génétique a été terminé, le risque de cancer peut être assigné pour le patient et sa famille. Quand une mutation a été identifiée, les tests génétiques devraient être offerts à tous les membres de la famille. Ces personnes doivent subir chaque conseil génétique ainsi que les avantages et les inconvénients des tests peuvent être expliquées. Porteurs d'une mutation sont offerts surveillance intensive. Si aucune mutation est identifiée chez le patient, mais la famille répond aux critères d'Amsterdam le protocole pour le syndrome de Lynch, la surveillance doit observer. La mortalité par cancer colorectal de ce groupe a été montré pour être réduit par la coloscopie de dépistage (32). La plupart des autorités estiment que le risque de réduction de l'hystérectomie avec salpingo-ovariectomie bilatérale a un rôle dans la gestion de ces patients. Les cancers colorectaux dans le syndrome HNPCC ont une propension à se produire dans le côlon proximal, par rapport à des cas sporadiques, qui sont plus souvent distale. Colectomie prophylactique est recommandé aux patients atteints de HNPCC mutation dans laquelle les polypes sont identifiés à un jeune âge et aussi aux patients qui ont des polypes qui sont l'instabilité positif ou ces personnes qui sont incapables de se soumettre à une surveillance régulière microsatellite. La chimioprévention utilisant des agents anti-inflammatoires non stéroïdiens dans ce groupe de patients pour prévenir les polypes du côlon est actuellement à l'étude, et des données devrait être à venir. Si ces patients sont à l'étude pour une autre raison, il faudrait envisager de counseling sur les risques et les avantages d'une hystérectomie totale avec salpingo-ovariectomie bilatérale possibles.
La surveillance de HNPCC (30),(33):
| Tumor Site | Screening Modality (%) |
| Colon | Colonoscopy every 1-2 years starting at ages 25-30 |
| Endometrium | Ultrasonography every 1-2 years starting at ages 30-35; consider endometrial biopsy every 1-2 years |
| Ovary | Ultrasonography and CA 125 annually, starting at ages 30-35 |
| Upper urinary tract | Urine cytology every 1-2 years starting at ages 30-35 |
| Stomach | Gastroscopy every 1-2 years starting at ages 30-35, (when upper GI cancer occurs two or more times in a family) |
GI, gastrointestinal
Syndrome de Li-Fraumeni
Le syndrome de Li-Fraumeni (LFS) est associée à des mutations germinales du gène suppresseur de tumeur TP53 , qui provoquent une prédisposition très pénétrant à développer des sarcomes ainsi que du sein précoce, cerveau, surrénale, et d'autres cancers. Les personnes atteintes de LFS ont un risque absolu de développer un cancer d'environ 50% par l'âge de 30 ans, et 90% en 60 ans (34). Compte EPA pour un maximum de 1% des cancers du sein aux États-Unis; ces cancers peuvent avoir un phénotype caractéristique, exprimant des récepteurs hormonaux et HER2/neu. La fréquence des mutations de novo est estimée à 7% à 20%, de sorte que TP53 test peut être approprié pour certains patients en l'absence d'antécédents de cancer de la famille (34).
Syndrome de Cowden
Le syndrome de Cowden (CS) transmet une susceptibilité accrue au cancer du sein, de la thyroïde, de l'endomètre et du côlon, et hamartomas bénignes touchant plusieurs organes. Il s'agit d'une affection autosomique dominante associée à des mutations dans le gène suppresseur de tumeur PTEN. Cancers gynécologiques sont fréquents avec un risque de 5-10% des cancers de l'endomètre et le risque de 25-50% des cancers du sein (42). Manifestations extérieures comprennent une macrocéphalie et papules papillomateuses sur le visage, la muqueuse buccale, linguale, et les extrémités, qui sont presque omniprésent par l'âge de 30 ans. Le risque de cancer du sein à vie est de 25-50%, avec un âge moyen de diagnostic des 38-46 ans (35). Le risque à vie de cancer de l'endomètre est de 5-10% (35). Mutation germinale dans le gène PTEN gène sont identifiées dans environ 80% des patients répondant aux critères diagnostiques cliniques, près de la moitié d'entre eux n'ont pas d'antécédents familiaux de CS. La fréquence déclarée de CS est de 1 sur 200.000, mais il est probablement sous-diagnostiqué (35). Récurrentes, plusieurs polypes de l'endomètre laissent présager un risque élevé de cancer de l'endomètre chez les femmes avec CS. Surveillance de malignité et l'examen de l'hystérectomie après la maternité est terminé est justifiée (42).
Syndrome du cancer gastrique diffus héréditaire de
Syndrome de cancer gastrique diffus héréditaire de est associée à des mutations dans le CDH1 gène et se caractérise par un risque à vie de 80% de développer un cancer gastrique (36). Les femmes ayant CDH1 mutations ont jusqu'à un risque à vie de 60% de développer un cancer du sein lobulaire, mais la prévalence de CDH1 mutations chez les femmes sélectionnées pour l'âge précoce de survenue ou des antécédents familiaux de cancer du sein lobulaire est faible, d'environ 1% (36). Parmi les familles avec gastrique diffus et le cancer du sein lobulaire, CDH1 mutations sont identifiées dans 25-50% (36).
Syndrome de Peutz-Jeghers
Le syndrome de Peutz-Jeghers (PJS), résultant de modifications dans le STK11 gène, dispose polypes hamartomateuses tout le tractus gastro-intestinal, conduisant à une invagination intestinale et une occlusion intestinale. STK11 1 mutations véhiculent des risques accrus pour le sein, de l'ovaire, de l'utérus, du pancréas, de l'estomac et du côlon le cancer, en plus de sexe tumeurs bénignes de la moelle. Le risque de cancer du sein atteint 8% par âge de 40 ans et 32% à l'âge 60 ans. La moitié des STK11 porteurs de la mutation n'ont pas d'antécédents familiaux de PJS (37).
Neurofibromatose-1
Neurofibromatose-1 (NF-1) Caractéristique neurofibromes cutanés et périphériques malignes des tumeurs des gaines nerveuses, les troubles d'apprentissage, et une prévalence de 1 sur 3000 naissances vivantes (38). Une étude récente des femmes à la norme NF-1 a rapporté risque multiplié par cinq de développer un cancer du sein avant l'âge de 50 ans et 3 - à 4 fois plus de risque de cancer à vie (38).
Norme d'avancement du traitement du cancer
Comme le dépistage des cancers héréditaires est devenue plus facilement accessible, de nombreuses questions entourant la responsabilité, la gestion des risques et la sécurité des patients ont vu le jour. Comme dans toutes les questions médico-légales, ces sujets de préoccupation concernent généralement la norme de soin, de documentation, de consentement, les attentes des patients et le suivi. Beaucoup de fournisseurs estiment que le dépistage du cancer héréditaire n'est pas la norme de diligence dans le bureau de soins primaires. Cependant, il ya trois points qui sont très importants à retenir. C'est la norme de diligence pour obtenir une histoire complète et détaillée la famille et le mettre à jour sur une base régulière. C'est la norme des soins à donner aux patients une information appropriée sur la base que l'histoire de la famille afin qu'ils puissent prendre des décisions éclairées au sujet de leurs soins médicaux. Enfin, il est la norme de diligence à l'enregistrement à fond et complètement tout ce qui était discutée avec le patient. Si vous adhérez à ces trois points, il semblerait que le dépistage du cancer héréditaire est, en fait, une norme de soins.
Documentation: une fois que vous avez identifié une personne qui répond aux critères pour les tests génétiques, combien de documents sont nécessaires? Est-il suffisant d'avoir votre état de la note: «les informations sur les tests génétiques donné" ou "brochure donnée?" mais il est agréable de voir votre plan documenté, il est beaucoup plus important de voir le raisonnement derrière le plan. Dans ce cas, une note élargi tels que:. "Basée sur les antécédents familiaux, les tests génétiques recommandé patient comprenne qu'il ya si le test est positif représente une augmentation substantielle du risque de cancer des ovaires et / ou un cancer du sein ou [le cancer du syndrome de Lynch particulier vous êtes dépistage de] ». Même si nous savons que nous avons discuté des risques de cancer, le patient peut facilement contredire ce qui n'est pas documenté dans leur dossier. Les patients peuvent faire valoir que s'ils avaient compris leurs risques, ils seraient, bien sûr, ont consenti à l'épreuve. Intégrer une sorte de système de suivi dans votre bureau est prudent. Cela peut permettre pour vous de suivi avec un patient après qu'elle a été renvoyée pour le conseil génétique. Sans ce type de suivi et de suivi, une question troublante peut être soulevée: «si vous senti que c'était assez important pour le patient d'avoir ce test, pourquoi n'était-elle pas assez important pour vous de voir si le test a été fait"
Consentement éclairé: consentement éclairé ou le refus éclairé doivent être abordées lors de l'examen des évaluations des risques héréditaires. En règle générale, le consentement éclairé a traité seulement d'informer les patients des risques liés à des procédures invasives. Cependant, il ya eu une expansion de ce consentement éclairé adéquat comprend. Dans le cadre du consentement éclairé adéquat On nous demande maintenant de donner à toutes les options de traitement, ainsi que les risques et les avantages de chaque option. Par conséquent, si nous ne donnons pas les patients appropriés la possibilité de tests génétiques (avec ses risques et bénéfices), nous pouvons être jugée négligente sur la base d'un consentement devrait y avoir un événement indésirable. C'est là que le refus éclairé peut entrer en jeu, si un patient ne veut pas faire ce que le fournisseur juge approprié, ou n'a pas suivi d'un conseil génétique référence lorsque vous lui parlez à l'un, de documenter leur refus, ou l'absence de suivi -up, peut finalement être plus important que de documenter leur consentement. Documents de refus éclairé que le médecin a fait ce qui est prudent et que c'est le choix du patient de ne pas suivre à travers. De nombreux États ont un élément de négligence contributive, et cela peut aller plus loin et de documenter la raison du refus du patient, la peur du résultat du test, le refus de faire quoi que ce soit sur le résultat, ou des raisons financières peut faire partie de la décision d'un patient à refuser le test.
À l'heure actuelle, l'une des principales causes de cas de faute professionnelle porte sur des questions présentant un cancer du sein. En règle générale, les allégations comprennent à la fois un diagnostic tardif et l'échec à diagnostiquer. Nous assistons aujourd'hui à une nouvelle allégation qui est considéré comme un échec de notre «devoir d'informer» ou «devoir d'avertir". Cela se rapporte à l'impossibilité d'identifier un patient à risque de cancer héréditaire de sorte que la surveillance accrue a pu être mis en uvre pour diagnostiquer le cancer plus tôt ou que la chirurgie de réduction des risques ou prophylactique auraient pu être réalisée. Ces types de cas, il sera très difficile, voire impossible, pour défendre sans documentation appropriée, y compris la documentation du refus du test d'un patient, et la documentation de l'explication des risques très spécifiques du cancer.
Résumé
Plus de 300.000 femmes américaines sont estimés à porter un risque élevé de développer hérité cancer du sein et de l'ovaire. Un plus large éventail de tests génétiques est disponible pour définir le risque du patient, mais les résultats encore peut s'avérer concluantes ou compliqué à interpréter. Cancer professionnels de la génétique offrent évaluation complète des risques, des conseils et des recommandations de gestion pour les patients et les familles ayant une prédisposition héréditaire pour le développement de cancers du sein et de l'ovaire. En tant que spécialistes de la santé des femmes, l'évaluation du risque héréditaire est de notre responsabilité. Une histoire familiale de cancer ciblée devrait faire partie de l'évaluation initiale de tous les patients. Pedigrees maternelle et paternelle doivent être construits de façon à inclure au moins trois générations (, ses parents du patient, et ses grands-générations). Les informations concernant l'âge du membre de la famille au moment du diagnostic et des détails supplémentaires telles que le cancer du sein dans les deux seins sont très utiles. Mises à jour de l'histoire de la famille doivent être posées sur les examens annuels à cause des ajouts et les nouveaux diagnostics peuvent modifier la recommandation initiale concernant les tests génétiques. Une évaluation du risque génétique est recommandé pour les patients avec un supérieur à une participation d'environ 20-25% de chance d'avoir une prédisposition héréditaire au cancer du sein et le cancer de l'ovaire. Les femmes ayant BRCA1 ou BRCA2 mutations devraient être offerts réduction des risques ovariectomie par l'âge de 40 ans ou lorsque de procréer est terminée. Pour une réduction des risques salpingo-ovariectomie bilatérale, tout le tissu des ovaires et des trompes de Fallope doit être supprimée. Visualisation complète des surfaces péritonéales avec lavage du bassin doit être effectuée. Complete sectionnement, série des ovaires et des trompes de Fallope est nécessaire, à l'examen microscopique du cancer occulte.
Références
- Robson ME, Boyd J, Borgen PI, et al. Hereditary-breast cancer. Curr Probl Surg 2001;38(6):387-480
- Risch HA, McLaughlin JR, Cole DC, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst 2006;98(23):1694-1706
- Dominguez FJ, Jones JL, Zabicki K, et al. Prevalence of hereditary breast/ovarian carcinoma risk in patients with a personal history of breast or ovarian carcinoma in a mammography population. Cancer 2005;104:1849-1853
- National Comprehensive Cancer Network. NCCN Guidelines: Genetic/Familial High-Risk Assessment: Breast and Ovarian Cancer. Available at: http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf Accessed 7 December 2012
- Reedy M, Gallion H, Fowler JM, et al. Contribution of BRCA1 and BRCA2 to familial ovarian cancer: a gynecologic oncology group study. Gynecol Oncol 2002;85:255-259
- Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006;25:5864-5874. (Level III)
- Venkitaramen AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002;108:171-182. (Level III)
- Karlan BY, Berchuck A, Mutch D. The role of genetic testing for cancer susceptibility in gynecologic practice. Obstet Gynecol 2007;110:155-167
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003;72:1117-1130. (Level III)
- Metcalfe KA, Lynch HT, Ghadirian P, et al. The risk of ovarian cancer after breast cancer in BRCA1 and BRCA2 carriers. Gynecol Oncol 2005;96:222-226. (Level II-3)
- Lakhani SR, Manek S, Penault-Llorca F, et al. Pathology of ovarian cancer in BRCA1 and BRCA2 carriers. Clin Cancer Res 2004;10:2473-2481. (Level II-3)
- Levine DA, Argenta PA, Yee CJ, et al. Fallopian tube and primary peritoneal carcinomas associated with BRCA mutations. J Clin Oncol 2003;21:4227-4227. (Level III)
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin. Hereditary breast and ovarian cancer syndrome. Number 103, April 2009
- National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast and ovarian. NCCN Clinical Practice Guidelines in Oncology. V.1.2008. Fort Washington (PA): NCCN; 2008. (Level III)
- Olivier RI, Lubsen-Brandsama MA, Verhoef S, et al. CA 125 and transvaginal ultrasound monitoring in high-risk women cannot prevent the diagnosis of advanced ovarian cancer. Gynecol Oncol 2006;100:20-26. (Level II-3)
- Whittemore AS, Balise RR, Pharoah PD, et al. Oral contraceptive use and ovarian cancer risk among carriers of BRCA1 or BRCA2 mutations. Br J Cancer 2004;91:1911-1915
- Rebbeck TR, Lynch HT, Neuhausen SL, et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 2002;346:1616-1622
- King MC, Marks JH, Mandell JB. The New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003;302:643-646
- Kauff ND, Satagopan JM, Robson ME, et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med 2002;346:1609-1615
- Karlan BY. Defining cancer risks for BRCA germline mutation carriers: implications for surgical prophylaxis. Gynecol Oncol 2004;92:519-520. (Level III)
- Lu KH, Kauff ND. Does a BRCA mutation plus tamoxifen equal hysterectomy? Gynecol Oncol 2007;104:3-4. (Level III)
- Satagopan JM, Boyd J, Kauff ND, et al. Ovarian cancer risk in Ashkenazi Jewish carriers of BRCA1 and BRCA2 mutations. Clin Cancer Res 2002;8:3776-3781. (Level II-3)
- Eisen A, Luinski J, Klijn J, et al. Breast cancer risk following bilateral oophorectomy in BRCA1 and BRCA2 mutation carriers: an international case-control study. J Clin Oncol 2005;23:7491-7496
- Walsh T, Casadei S, Coats KH, et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA2006;295:1379-1388. (Level III)
- Rodriquez E, Domchek SM. The prevention of hereditary breast cancer. Semin Oncol 2007;34:401-405. (Level III)
- Kauff ND, Mitra N, Robson ME, et al. Risk of ovarian cancer in BRCA1 and BRCA2 mutation-negative hereditary breast cancer families. J Natl Cancer Inst 2005;97:1382-1384. (Level II-3)
- Offit K, Kohut K, Claggett B, et al. Cancer genetic testing and assisted reproduction. J Clin Oncol 2006;24:4775-4782
- Offit K, Sagi M, Hurley K. Preimplantation genetic diagnosis for cancer syndromes: a new challenge for preventive medicine. JAMA 2006;296:2727-2730
- Lynch HT, Wastson P, Shaw TG, et al. Clinical impact of molecular and genetic diagnosis, genetic counseling, and management of hereditary cancer. Part I: Studies of cancer in families. Cancer 1999;86:2449-2456
- Hendriks YM, de Jong AE, Morreau H, et al. Diagnostic approach and management of Lynch syndrome (hereditary nonpolyposis colorectal carcinomas): a guide for clinicians. CA Cancer J Clin 2006;56:213-225
- Syngal S, Fox EA, Eng C, et al. Sensitivity and Specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet 2000;37:641-645
- Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000;118:829-834
- Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res2006;66:7810-7817
- Gonzalez KD, Buzin CH, Noltner KA, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Gent 2009;46(10):689-693
- Zhou XP, Waite KA, Pilarski R, et al. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN/ protein and dysregulation of the phosphoinsositol-3-kinase/Akt pathway. Am J Hum Genet 2003;73(2):404-411
- Schrader KA, Masciari S, Boyd N, et al. Germline mutations in CDH1 are infrequent in women with early-onset or familial lobular breast cancer. J Med Gent 2011;48(1):64-68
- Lim W, Olschwang S, Keller JJ, et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 2004;126(7):1788-1794
- Sharif S, Moran A, Huson SM, et al. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet 2007;44(8):481-484
- Pearlman MD. Ideal risk reduction management for women with BRCA gene mutations. Obstet Gynecol 2013;121:4-6
- Kwon JS, Tinker A, Pansegrau G, et al. Prophylactic salpingectomy and delayed oophorectomy as an alternative for BRCA mutation carriers. Obstet Gynecol 2013;121:14-24
- Mecalfe KA, Birenbaum-Carmeli D, Lubinski J, et al. International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int J Cancer 2008;122:2017-2022
- Kalin A, Merideth MA, Regier DS, et al. Management of reproductive health in Cowden Syndrome complicated by endometrial polyps and breast cancer. Obstet Gynecol 2013;121:461-464
Publié: 25 April 2013
Dedicated to Women's and Children's Well-being and Health Care Worldwide
www.womenshealthsection.com